Abstract
The cricket industry in South Korea has experienced significant annual growth, supported by advanced technology and government programs. These advantages have enabled the insect industry to establish mass rearing methods; however, these methods are vulnerable to epizootic diseases, particularly those caused by entomopathogens. In this study, we collected crickets from various farms and analyzed their virome composition using nine different PCR primer sets for cricket viruses. While volvovirus was not detected at all cricket farms, it was the only virus found. Consequently, the focus of this study was on the genomic analysis and molecular characterization of a novel strain of volvovirus identified in Gryllus bimaculatus in South Korea, designated as GbVVV-KR. The complete genome sequence of GbVVV-KR was obtained through Sanger sequencing using degenerate primers, revealing a circular single-stranded DNA virus with a genome size of 2,515 nucleotides. The genomic analysis showed variations, including single-nucleotide polymorphisms and insertions/deletions, with a notable mutation occurring at the 112th codon (GAA) in ORF3, which resulted in a stop codon (TAA). Additionally, when assessing the prevalence of GbVVV-KR, the virus was detected in 8 out of 11 farms across South Korea, with varying viral loads observed among the farms. This study provides valuable insight into the genetic characteristics, phylogeny and prevalence of GbVVV-KR, and contributes to a broader understanding of entomopathogenic viruses in crickets.
1 Introduction
Among the edible insects currently approved by the Korea Food and Drug Administration, two-spotted crickets (Gryllus bimaculatus) are particularly popular due to their easy husbandry and high nutritional value. As a result, crickets are utilized not only as supplements and feed for various reptiles and fish but also as food for human consumption in many countries (Han et al., 2017). The cricket industry in South Korea has seen significant growth in recent years, with the latest report from the Rural Development Administration indicating that approximately 2.7 million crickets are sold annually (R.D.A., 2022). Although the number of cricket farms has decreased lately, cricket sales have risen owing to mass production techniques, including vertical farming and technological advancements. However, these mass-rearing systems may be vulnerable to epizootic diseases (Eilenberg et al., 2015; Maciel-Vergara and Ros, 2017). Despite the occurrence of insect epidemics since the start of the insect industry, there is limited research on entomopathogenic microorganisms that adversely affect farmed insects (Bahadur, 2018; Bertola and Mutinelli, 2021).
Disease management protocols for industrial insects are necessary, prompting efforts to characterize newly identified viruses (de Miranda et al., 2021a). Research on cricket viruses has revealed that crickets can be infected by a variety of viruses. Notable examples include Acheta domesticus densovirus (AdDV) (Szelei et al., 2011), Acheta domesticus volvovirus (AdVVV) (Pham et al., 2013a,b), Acheta domesticus iflavirus (AdIV) (de Miranda et al., 2021b), Acheta domesticus mini ambidensovirus (AdMADV) (Pham et al., 2013c), Gryllus bimaculatus nudivirus (GbNV) (Wang et al., 2007), invertebrate iridovirus 6 (IIV-6) (Papp and Marschang, 2019), cricket paralysis virus (Kerr et al., 2018; Valles and Chen, 2006), slow bee paralysis virus (de Miranda et al., 2010), and Acheta domesticus segmented densovirus (AdSDV) (PeÌnzes et al., 2023). In Sweden, the diversity and quantity of viruses affecting Acheta domesticus varied depending on the life stages and retailers involved (de Miranda et al., 2021a). Among the virus groups, AdDV and IIV-6 were fairly common, and they were even detected in frass (de Miranda et al., 2021a). Therefore, regular viral screening and monitoring should be implemented for cricket species used in each country.
Regarding cricket volvoviruses, five full genome sequences are registered in the GenBank (Homchan and Gupta, 2024; Pham et al., 2013a,b). So far, it has been shown that cricket volvovirus infects A. domesticus and G. assimilis (Jamaican field cricket) (Rosario et al., 2018). The volvovirus is characterized by a circular single-stranded DNA genome, measuring 2,516â2,517 nucleotides (nt) in size. Additionally, the volvovirus features a putative nonanucleotide origin of replication (5â²-TAGTATTAC-3â²), which forms a hairpin structure at the beginning of the genome (Osinga et al., 1982). The genome of the cricket volvovirus contains four open reading frames (ORFs) (Tijssen et al., 2016).
Here, we present the first identification of a volvovirus-infected G. bimaculatus from cricket farms in South Korea, referred to as GbVVV-KR. Because of the size of the genome and high similarity between virus sequences, the sequences were obtained by Sanger sequencing with degenerate primers. Phylogenetic analysis revealed the evolutionary relationships between known volvovirus isolates and the newly discovered GbVVV-KR. To investigate the potential spread of this virus across the country, crickets were collected from 11 different farms in South Korea. The prevalence of the virus and viral loads were determined using conventional PCR and quantitative real-time PCR (qPCR). The genomic analyses and molecular studies provide a valuable resource for those seeking a better understanding of cricket volvovirus and its associated viral community.
2 Materials and Methods
Genome sequencing and assembly
To examine domestic cricket viruses, four farms were screened using nine previously reported primers (de Miranda et al., 2021a) (Supplementary Table S1). Total DNA and RNA were extracted from whole-body homogenates using the Clear-S⢠DNA/RNA extraction kit (Invirustech, Gwangju-si, South Korea), based on the manufacturerâs instructions. For each sample, three crickets were pooled per tube. For RNA viruses, total RNA was converted into cDNA using ReverTra Ace⢠qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan). For DNA viruses, PCR was performed directly on the total DNA extracts (Supplementary Figure S1). All PCR reactions were performed using the following thermocycling conditions: initial denaturation at 95 °C for 3 min; 35 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min.
To develop primers targeting volvovirus, we aligned publicly available volvovirus sequences from the National Center for Biotechnology Information (NCBI). The five volvovirus genome sequences (accession numbers KC543331.1, KC794540.1, KC794539.1, MH545526.1, and MW288623.1) were obtained from the NCBI database.
The genome was amplified by PCR into the following four regions using primer sets 1â4 from Table 1: 1,004 bp for the 496â1,499 nt region; 1,001 bp for the 1,469â2,469 nt region; 863 bp for the 2,282â629 nt region, and 792 bp for the 853â1,644 nt region. KOD FX neo (Toyobo) was used for all PCR reactions run on a C1000 Touch Thermal Cycler (Bio-Rad, Hercules, CA, USA). The PCR conditions included 35 cycles at 94 °C for 30 s, 55 °C for 25 s, and 68 °C for 50 s. The PCR products were purified using Clear-S⢠PCR/Gel DNA fragment purification kit (Invirustech, Gwangju, South Korea). Sanger sequencing was performed by Bioneer (Daejeon, South Korea).
To rule out the possibility of PCR polymerase and sequencing errors, the 1,469â2,469 nt region (1,001 bp) was re-examined using primer set 3 with a different polymerase, CloneAmp HiFi PCR Premix (Takara Bio Inc., Kusatsu, Japan). The PCR conditions were 94 °C for 20 s, 60 °C for 15 s, and 68 °C for 15 s for 35 cycles. Sequencing was performed using the same method as above. The sequences obtained from Sanger sequencing were assembled as a whole circular genome using SnapGene software (https://www.snapgene.com/).
Phylogenetic tree
To explore the evolutionary relationships among volvoviruses, five complete volvovirus genome sequences were collected from the NCBI database (Table 2) along with GbVVV-KR. Nucleotide sequences were aligned, and phylogenetic analyses were conducted using MEGA 11 software (http://www.megasoftware.net/).
The tree was inferred using the maximum likelihood method with 1,000 bootstrap replicates, and the GTR + G model was selected as the best-fit substitution model. Bootstrap values above 70% were considered significant.
Pairwise genetic distances (p-distances) among complete genome sequences of the volvoviruses were computed in MEGA11. The p-distance was calculated as the number of differing nucleotide sites divided by the total number of compared sites, excluding positions with gaps or missing data (pairwise deletion option). The resulting distance matrix was visualized as a heatmap using the seaborn package in Python.
ORF identification and analysis
Potential ORFs were predicted in the assembled whole-genome sequence of GbVVV-KR using the NCBI ORFfinder (https://www.ncbi.nlm.nih.gov/orfinder/) with the option of methionine-initiated ORFs encoding more than 30 amino acids. Identified ORFs were annotated for known functions or motifs using the NCBI BLASTx and CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Relative viral abundance across South Korea
Crickets (G. bimaculatus) were collected from 11 farms across the country. For DNA extraction, three crickets were placed in a single tube, and total DNA was extracted using the Clear-S Quick DNA extraction kit (IVT3002, Invirustech Co., South Korea) according to the manufacturerâs instructions. The concentration of extracted DNA was measured using OPTIZEN⢠NanoQ Plus (K LAB, Daejeon, South Korea). To visualize differences in band intensity corresponding to infection levels, endpoint PCR was performed using the GbVVV_3 F and GbVVV_2 R primers (Table 1). To examine the interindividual transmission of the volvovirus, a specific farm was selected, and infection levels were analyzed using 12 individual crickets. Each cricket was placed separately in a tube, and total DNA was extracted from each one.
Quantitative PCR
To determine the presence of GbVVV in the samples, qPCR was performed using the primer sets listed in Supplementary Table S2, along with PowerUp⢠SYBR⢠Green Master Mix (A25741, Thermo Fisher Scientific, Waltham, MA, USA). The qPCR thermal cycling conditions were as follows: 95 °C for 15 s, 60 °C for 15 s and 72 °C for 60 s, for a total of 40 cycles. Reactions were carried out on a Bio-Rad CFX96 system, and the results were analyzed using the Bio-Rad CFX Maestro software (Bio-Rad).
GbVVV is a single-stranded DNA (ssDNA) virus; however, amplification was performed directly from total DNA without prior second-strand synthesis. During the initial denaturation step, the viral ssDNA was rendered accessible for primer annealing and polymerase extension. Melting curve analysis was conducted for each primer set to confirm specificity and ensure that no off-target amplification or primer-dimer formation occurred.
G. bimaculatus β-actin was used as the reference gene for normalization. Although two reference genes were initially considered, only one primer set for β-actin was used in the final analysis because of its consistent amplification efficiency. ÎCt values were calculated by subtracting the Ct value of the β-actin gene from the Ct value of GbVVV-KR ORF2. The resulting Ct values were interpreted as exploratory indicators of relative viral DNA levels rather than as absolute quantification.
3 Results
Construction of the GbVVV-KR genome and structure features
A complete genomic sequence (2,515 nt) was obtained by assembling the overlapping portions of the Sanger sequencing results. BLAST analysis of the sequence in the NCBI database showed 100% sequence coverage and identities between 97.74 to 98.13% at the nucleotide level, identifying the sequence as a volvovirus (Supplementary Table S3).

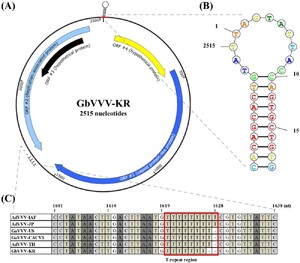
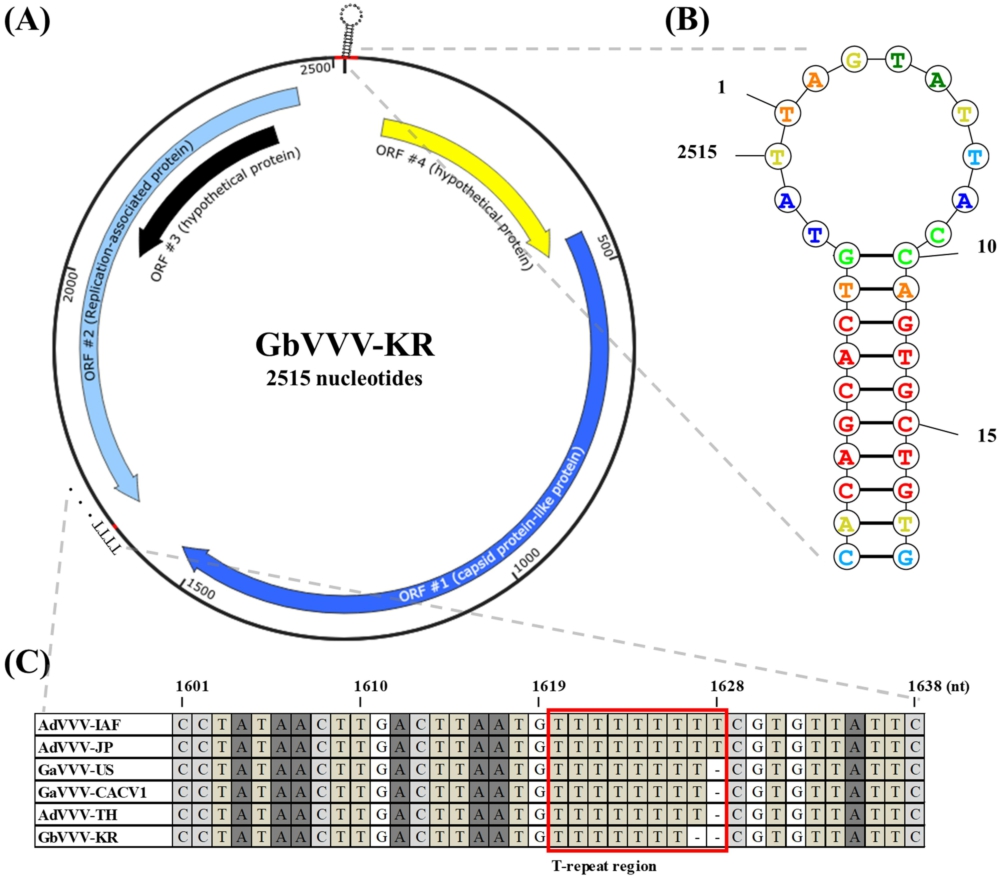
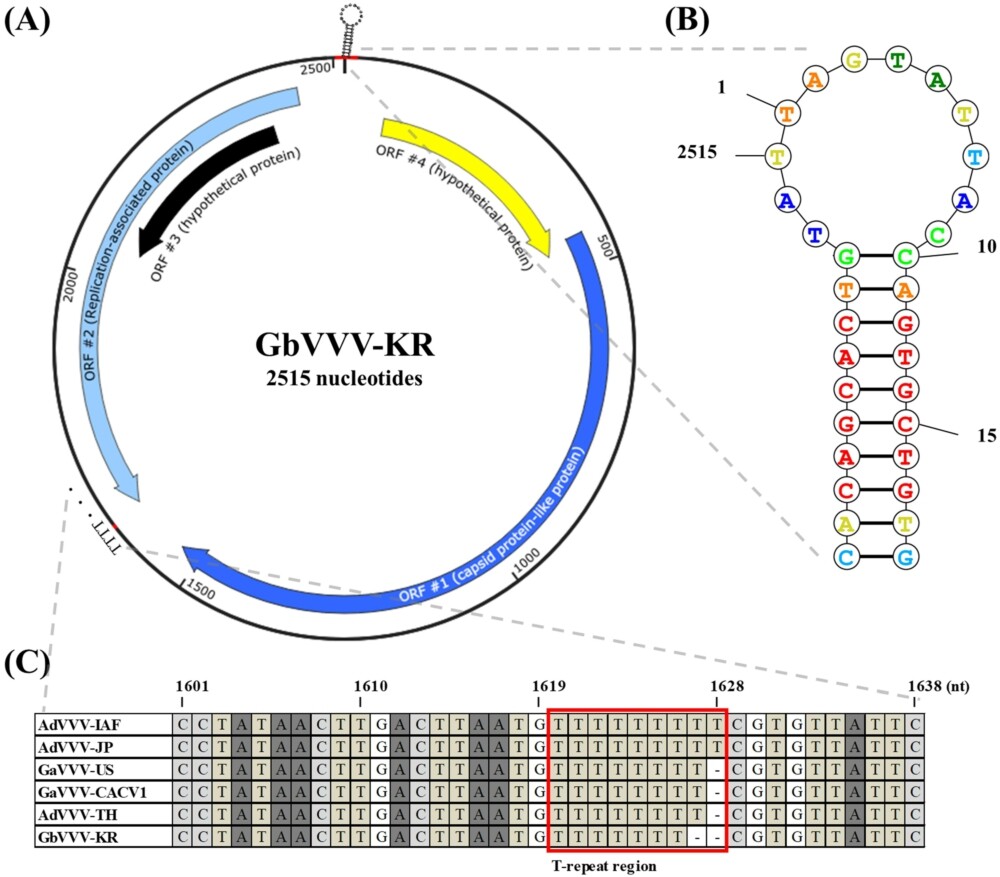
GbVVV genome map isolated from South Korea. (A) There are four ORFs among the 2,515 nucleotides in the GbVVV-KR. ORF4 (starting at 70 nt) and ORF1 (starting at 447 nt) are located in the sense orientation. ORF2 (starting at 2,443 nt) and ORF3 (starting at 2,391 nt) are located in an antisense orientation. (B) The structure of the putative nonanucleotide origin of replication (5â²-TAGTATTAC-3â²) is enlarged, and the probability values are indicated by color. (C) The difference in the length of the sequence of each genome occurs from the T-repeat region (1,620 nt). Detailed information on the virus abbreviations is provided in Table 2.
Citation: Journal of Insects as Food and Feed 12, 5 (2026) ; 10.1163/23524588-bja10311
GbVVV-KR was predicted to have four ORFs (Figure 1A and Supplementary Table S4). ORF1 was annotated as a structural protein-related gene (accession number QQM18919.1, e-value 0.0) and ORF2 as a replication-related gene (accession number YP_007878130.1, e-value 0.0). The functions of ORF3 and ORF4, which are hypothetical proteins, are unknown (Figure 1A). GbVVV-KR contained a nonamer sequence (underlined) located at the start of the genome within a stem-loop structure (5â²-CACAGCACTGTATTAGTATTACCAGTGCTGTG-3â²) (1â19 nt) and at the end of the genome (2,503â2,515 nt) (Figures 1A,B). This sequence forms a hairpin because of the stem-loop structure, which was also identified in GbVVV-KR (Figure 1B). These results indicate that GbVVV-KR contains a circular genome and a nonanucleotide motif in the stem-loop sequence.
The genome size of volvoviruses varied slightly between isolates. AdVVV-IAF and AdVVV-JP were identical at 2,517 bp. GaVVV-US, GaVVV-CACV1, and AdVVV-TH were one bp shorter than AdVVV-IAF at 2,516 bp. GbVVV-KR was one bp shorter than GaVVV-US at 2,515 bp (Supplementary Figure S2). The difference in genome size between the isolates was attributed to a base drop in the T-repeat region beginning at nucleotide 1,620 (Figure 1C). This T-repeat region is located at the end of the ORF2, but does not belong in any ORF sites (Figure 1C).
Phylogenetic analysis
A rooted phylogenetic tree was constructed to examine the evolutionary relationships among the six volvovirus isolates (Figure 2A). Due to the high sequence similarity among the isolates (>95% nucleotide identity), the tree displayed very short branch lengths, indicating limited genetic divergence. Within the volvovirus group, the isolates AdVVV-IAF, AdVVV-JP, GaVVV-US, and GaVVV-CACV1 clustered together, while AdVVV-TH and GbVVV-KR formed separate subgroups (Figure 2A).
Phylogenetic relationships and pairwise genetic distances among volvoviruses. (A) The phylogeny was reconstructed using the maximum likelihood method under the GTR + G substitution model, based on whole-genome nucleotide alignments. Bootstrap support values (>70%, 1,000 replicates) are indicated at the major nodes. (B) Pairwise genetic distances among six volvovirus isolates, calculated using the Kimura 2-parameter (K2P) model in MEGA11. Values represent substitutions per site. The color gradient indicates relative divergence, with black corresponding to more similar pairs and white corresponding to more divergent pairs.
Citation: Journal of Insects as Food and Feed 12, 5 (2026) ; 10.1163/23524588-bja10311
To further resolve these relationships, pairwise genetic distances (PGDs) among the six isolates were calculated using the Kimura 2-parameter (K2P) model (Figure 2B). PGD analysis revealed that GbVVV-KR exhibited the greatest genetic distances relative to the other isolates, with values ranging from 0.0185 to 0.0222 substitutions per site, which is consistent with its more divergent position on the phylogenetic tree. In contrast, the group composed of AdVVV-IAF, AdVVV-JP, GaVVV-US, and GaVVV-CACV1 showed relatively low genetic distances among one another (0.0020â0.0092), indicating close evolutionary relatedness (Figure 2B). AdVVV-TH exhibited intermediate genetic distances, showing greater divergence compared to the four closely related isolates but having similar or slightly smaller genetic distances to GbVVV-KR.
These analyses together highlight two notable findings: (1) volvoviruses classified within the same species (AdVVV) can be differentiated by geographic origin and (2) the two most recently identified isolates (AdVVV-TH and GbVVV-KR) exhibited greater genetic divergence from the other four.
SNP-based genomic analysis and comparison of the ORFs
Because AdVVV-IAF was identified first, we used it as a reference sequence to compare other volvovirus sequences. ORF1, a hypothetical protein/capsid protein-like protein, appeared to be the most diversified sequence compared with the other ORFs: 18 polymorphic sites were found in AdVVV-JP, 5 in GaVVV-US, 6 in GaVVV-CACV1, 38 in AdVVV-TH, and 31 in GbVVV-KR vs. AdVVV-IAF (Table 3). With respect to the amino acid sequence, nonsynonymous mutations were identified in ORF1: 14 mutations in AdVVV-JP, 5 mutations in GaVVV-US and GaVVV-CACV1, 34 mutations in AdVVV-TH, and 28 mutations in GbVVV-KR (Table 3 and Supplementary Figure S3A). ORF2 was highly conserved in most of the sequences (Table 3 and Supplementary Figure S3B), with a P-loop superfamily motif present within the sequence (Supplementary Figure S4). In ORF4, nonsynonymous mutations were present only in AdVVV-JP, AdVVV-TH, and GbVVV-KR, whereas the sequence was conserved in the other isolates (Table 3 and Supplementary Figure S3C).
Amino acid sequence alignment and SNP analysis of ORF3 for all reported volvoviruses and GbVVV-KR. (A) Schematic diagram indicating the location of the ORFs in the volvovirus, including the ORF3 of GbVVV-KR. (B) The volvovirus ORF3 amino acid sequence aligned with the Mega 11 program. Dots (.) indicate consensus sites, and red boxes highlight the mutation sites, indicating nonsynonymous SNPs and premature stop codons (*). AdVVV-IAF served as the reference sequence.
Citation: Journal of Insects as Food and Feed 12, 5 (2026) ; 10.1163/23524588-bja10311
Comparing GbVVV-KR to the other volvovirus genomes, the most significant alteration occurred in ORF3, which encodes a hypothetical protein with no functional prediction. A stop codon generated a truncated ORF in the SNP that codes for glutamate (GAA) into one that codes for the stop codon (TAA) (Figure 3). Because of this mutation, ORF3 became a partial ORF with 111Â aa. The size of ORF3 in other isolates was 207 aa. To exclude the possibility of technical errors, this mutation site and a T-repeat region were re-examined using a different primer set and a high-fidelity DNA polymerase, followed by Sanger sequencing. The results were consistent with the original sequence, confirming that the observed mutations were not artifacts.
Relative viral signal distribution across regions
To evaluate the prevalence of volvovirus, crickets were collected from 11 farms across South Korea, and their locations were mapped on a regional map (Figure 4A). The presence and level of the virus were confirmed through both conventional PCR and qPCR analyses (Figure 4B). Conventional PCR revealed clear bands in some farms with strong infection, while only faint or no bands were observed in farms with relatively weaker infection. For qPCR, the Ct values of the cricket reference gene ranged from 21.85 to 24.75, whereas virus-specific Ct values ranged from 28.57 to not detected. Notably, Farm I showed the strongest viral signal, with a ÎCt value of 3.89 (Figure 4B).
Exploratory relative viral signal analysis of cricket farms using qPCR. (A) Farm labels (AâK) represent different collection sites in South Korea. A map illustrates the geographic distribution of the 11 domestic farms where samples were collected. (B) qPCR analysis of pooled cricket samples (
Citation: Journal of Insects as Food and Feed 12, 5 (2026) ; 10.1163/23524588-bja10311
Since viral loads varied significantly between farms, we conducted further investigations to determine whether relative viral signals varied within a farm (Figure 4C). From 12 crickets from Farm A, individual viral loads ranged from 27.62 to 39.62. The two ÎCt values with the greatest difference were 8.76, which shows a relative difference in viral load of 433-fold.
Taken together, these results demonstrate that volvovirus prevalence and abundance varied substantially across farms and that notable heterogeneity was also present among individual crickets within the same farm. The ÎCt values presented here should be regarded as proxies of relative viral abundance rather than absolute viral loads.
4 Discussion
Crickets are raised around the world for food and various other purposes, and their importance and value continue to increase. However, mass mortality caused by microorganisms, particularly viruses, has been experienced in various cricket species. Continuous research on viruses, which pose a constant threat to the cricket industry, is essential. Here, we report the genomic characterization of a volvovirus infecting two-spotted crickets (G. bimaculatus), which are commonly farmed in South Korea. Genetic characteristics differ from previously known volvoviruses, including numerous nonsynonymous mutations and an early termination of ORF3 resulting from a stop codon mutation. This study summarized and comprehensively analyzed the volvoviruses discovered in crickets.
Based on our sequence analysis, we made several interesting observations. First, the volvovirus isolates exhibited comparable genome sizes ranging from 2,515 to 2,517 nt, with size variation attributed to a T-repeat region at 1,620 nt (Figure 1). Second, ORF2 (replication-associated protein), ORF3 (hypothetical protein), and ORF4 (hypothetical protein) are highly conserved among the volvoviruses, in contrast to ORF1 (capsid protein-like protein). The capsid is crucial for determining the viral surface morphology and plays an important role in identifying the virus isolates (Supplementary Figure S3) (Rosario et al., 2012). A comparison of ORF1 between the isolates AdVVV-TH and GbVVV-KR revealed a highly similar sequence pattern in terms of nucleotides and amino acids. These isolates carried a greater number of synonymous and nonsynonymous mutations compared with the other isolates (Table 3). Notably, 14 out of 31 polymorphic sites in GbVVV-KR were conserved, showing the same nonsynonymous mutations as AdVVV-TH, indicating a close genetic relationship and a potential evolutionary connection between them. Phylogenetic analysis and genetic diversity studies suggest that the genome sequences of GbVVV-KR and AdVVV-TH are comparable, despite being from different hosts (Figure 2). This finding implies that volvoviruses may also exist in other species. Single-stranded DNA viruses are highly susceptible to DNA damage and hypermutation, traits that may facilitate their genetic adaptation to diverse environments (Saini and Gordenin, 2020; Stedman, 2013). Interestingly, when examining the sizes of the ORFs, we found that the most distinct feature of GbVVV-KR was its shorter ORF3 compared with other volvoviruses (Figure 3). This truncation appears to result from an SNP that introduces a premature stop codon. Therefore, we propose that the cross-species transmission of volvoviruses may be driven by genetic variations in ORF1 (Cap) and the presence of an incomplete ORF3.
A decade ago, G. bimaculatus was introduced to South Korea for mass farming, and exchange systems between farms were established to meet consumer demands (Gupta et al., 2020). While this practice supported industry growth, it may also have facilitated viral dissemination among farms. Our qPCR survey revealed that volvovirus is present in most farms, with infection levels varying across regions and among individuals (Figure 4). Because covert or low-level infections are common in crickets (Duffield et al., 2021), the detection thresholds used here should be interpreted as relative rather than diagnostic. Volvoviruses have been reported to occur widely in several cricket species (de Miranda et al., 2021a), and viral interactions may influence host health and farming outcomes (Dolan et al., 2018; Domingo et al., 2012). Overall, these results indicate that volvovirus is already established in South Korean cricket farms. Therefore, continuous sequencing and monitoring of volvovirus and other cricket-associated viruses will be essential to improve our understanding of viral diversity in industrial insects and to support sustainable cricket farming.
Corresponding author; e-mail:Â jsyoon@jbnu.ac.kr
Conflict of interest
The authors declare that they have no competing financial interests.
Data availability
All volvovirus sequence data supporting the findings of this study have been deposited in GenBank. Gryllus bimaculatus volvovirus (GbVVV-KR) has been deposited under accession number PP275066. (https://www.ncbi.nlm.nih.gov/nuccore/2672398230).
Funding
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korean government (MEST) (No. 2022R1C1C1007039).
References
Bahadur, A.B., 2018. Entomopathogens: role of insect pest management in crops. Trends in Horticulture 1: 833. https://doi.org/10.24294/th.v1i4.833
Bertola, M. and Mutinelli, F., 2021. A Systematic Review on Viruses in Mass-Reared Edible Insect Species. Viruses 13: 2280. https://doi.org/10.3390/v13112280
de Miranda, J.R., Dainat, B., Locke, B., Cordoni, G., Berthoud, H., Gauthier, L., Neumann, P., Budge, G.E., Ball, B.V. and Stoltz, D.B., 2010. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.). Journal of General Virology 91: 2524-2530. https://doi.org/10.1099/vir.0.022434-0
de Miranda, J.R., Granberg, F., Low, M., Onorati, P., Semberg, E., Jansson, A. and Berggren, AÌ., 2021a. Virus Diversity and Loads in Crickets Reared for Feed: Implications for Husbandry. Frontiers in Veterinary Science 8: 642085. https://doi.org/10.3389/fvets.2021.642085
de Miranda, J.R., Granberg, F., Onorati, P., Jansson, A. and Berggren, AÌ., 2021b. Virus Prospecting in Crickets-Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus. Viruses 13: 364. https://doi.org/10.3390/v13030364
Dolan, P.T., Whitfield, Z.J. and Andino, R., 2018. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annual Review of Virology 5: 69-92. https://doi.org/10.1146/annurev-virology-101416-041718
Domingo, E., Sheldon, J. and Perales, C., 2012. Viral quasispecies evolution. Microbiology and Molecular Biology Reviews 76: 159-216. https://doi.org/10.1128/mmbr.05023-11
Duffield, K.R., Hunt, J., Sadd, B.M., Sakaluk, S.K., Oppert, B., Rosario, K., Behle, R.W. and Ramirez, J.L., 2021. Active and Covert Infections of Cricket Iridovirus and Acheta domesticus Densovirus in Reared Gryllodes sigillatus Crickets. Front Microbiol 12: 780796. https://doi.org/10.3389/fmicb.2021.780796
Eilenberg, J., Vlak, J.M., Nielsen-Leroux, C., Cappellozza, S. and Jensen, A.B., 2015. Diseases in insects produced for food and feed. Journal of Insects as Food and Feed 1: 87-102. https://doi.org/10.3920/jiff2014.0022
Gupta, Y.M., Tanasarnpaiboon, S., Buddhachat, K., Peyachoknagul, S., Inthim, P. and Homchan, S., 2020. Development of microsatellite markers for the house cricket, Acheta domesticus (Orthoptera: Gryllidae). Biodiversitas Journal of Biological Diversity 21. https://doi.org/10.13057/biodiv/d210921
Han, R., Shin, J.T., Kim, J., Choi, Y.S. and Kim, Y.W., 2017. An overview of the South Korean edible insect food industry: challenges and future pricing/promotion strategies. Entomological Research 47: 141-151. https://doi.org/10.1111/1748-5967.12230
Homchan, S. and Gupta, Y.M., 2024. New volvovirus isolate from Thai domesticated house cricket, Acheta domesticus (Orthoptera: Gryllidae). Journal of Insects as Food and Feed 10: 1897-1904. https://doi.org/10.1163/23524588-00001217
Kerr, C.H., Dalwadi, U., Scott, N.E., Yip, C.K., Foster, L.J. and Jan, E., 2018. Transmission of Cricket paralysis virus via exosome-like vesicles during infection of Drosophila cells. Scientific Reports 8: 17353. https://doi.org/10.1038/s41598-018-35717-5
Maciel-Vergara, G. and Ros, V.I.D., 2017. Viruses of insects reared for food and feed. Journal of Invertebrate Pathology 147: 60-75. https://doi.org/10.1016/j.jip.2017.01.013
Osinga, K.A., De Haan, M., Christianson, T. and Tabak, H.F., 1982. A nonanucleotide sequence involved in promotion of ribosomal RNA synthesis and RNA priming of DNA replication in yeast mitochondria. Nucleic Acids Research 10: 7993-8006. https://doi.org/10.1093/nar/https://doi.org/10.24.7993
Papp, T. and Marschang, R.E., 2019. Detection and characterization of invertebrate iridoviruses found in reptiles and prey insects in Europe over the past two decades. Viruses 11: 600. https://doi.org/10.3390/v11070600
PeÌnzes, J.J., Pham, H.T., Chipman, P., Smith, E.W., McKenna, R. and Tijssen, P., 2023. Bipartite genome and structural organization of the parvovirus Acheta domesticus segmented densovirus. Nature Communications 14: 3515. https://doi.org/10.1038/s41467-023-38875-x
Pham, H.T., Bergoin, M. and Tijssen, P., 2013a. Acheta domesticus volvovirus, a novel single-stranded circular DNA Virus of the house cricket. Genome Announcements 1: e0007913. https://doi.org/10.1128/genomeA.00079-13
Pham, H.T., Iwao, H., Bergoin, M. and Tijssen, P., 2013b. New volvovirus isolates from Acheta domesticus (Japan) and Gryllus assimilis (United States). Genome Announcements 1. https://doi.org/10.1128/genomeA.00328-13
Pham, H.T., Yu, Q., Bergoin, M. and Tijssen, P., 2013c. A novel ambisense densovirus, Acheta domesticus mini ambidensovirus, from crickets. Genome Announcements 1. https://doi.org/10.1128/genomeA.00914-13
R.D.A., 2022. 2022 Insect industry status survey results. Rural Development Administration, Seoul.
Rosario, K., Duffy, S. and Breitbart, M., 2012. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Archives of Virology 157: 1851-1871. https://doi.org/10.1007/s00705-012-1391-y
Rosario, K., Mettel, K.A., Benner, B.E., Johnson, R., Scott, C., Yusseff-Vanegas, S.Z., Baker, C.C.M., Cassill, D.L., Storer, C., Varsani, A. and Breitbart, M., 2018. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 6: e5761. https://doi.org/10.7717/peerj.5761
Saini, N. and Gordenin, D.A., 2020. Hypermutation in single-stranded DNA. DNA Repair (Amst) 91-92: 102868. https://doi.org/10.1016/j.dnarep.2020.102868
Stedman, K., 2013. Mechanisms for RNA capture by ssDNA viruses: grand theft RNA. Journal of Molecular Evolution 76: 359-364. https://doi.org/10.1007/s00239-013-9569-9
Szelei, J., Woodring, J., Goettel, M.S., Duke, G., Jousset, F.X., Liu, K.Y., Zadori, Z., Li, Y., Styer, E., Boucias, D.G., Kleespies, R.G., Bergoin, M. and Tijssen, P., 2011. Susceptibility of North-American and European crickets to Acheta domesticus densovirus (AdDNV) and associated epizootics. Journal of Invertebrate Pathology 106: 394-399. https://doi.org/10.1016/j.jip.20https://doi.org/10.12.009
Tijssen, P., PeÌnzes, J.J., Yu, Q., Pham, H.T. and Bergoin, M., 2016. Diversity of small, single-stranded DNA viruses of invertebrates and their chaotic evolutionary past. Journal of Invertebrate Pathology 140: 83-96. https://doi.org/10.1016/j.jip.2016.09.005
Valles, S.M. and Chen, Y., 2006. Serendipitous discovery of an RNA virus from the cricket, Acheta domesticus. Florida Entomologist 89: 282-283.
Wang, Y., Kleespies, R.G., Huger, A.M. and Jehle, J.A., 2007. The genome of Gryllus bimaculatus nudivirus indicates an ancient diversification of baculovirus-related nonoccluded nudiviruses of insects. Journal of Virology 81: 5395-5406. https://doi.org/10.1128/jvi.02781-06




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}