Abstract
The changing of microbiome could precede the development of coeliac disease (CeD). We compared the bacterial profile of microbiota of tissues collected simultaneously from the stomach and duodenum in newly diagnosed patients with CeD. Biopsies were collected from 60 children and adolescents aged 2-18 years: (1) 40 patients with CeD; (2) 20 children as control group. The evaluation of the bacterial microbiota was carried out by sequencing the V3-V4 regions of the 16S rRNA subunit, using next-generation sequencing (NGS). The composition of bacterial microbiota was correlated with clinical and blood parameters. The beta diversity analysis revealed a significant dissimilarity in the gastric samples between the CeD and control group (Bray-Curtis index, P = 0.008, and weighted UniFrac distance, P = 0.024). At L2 (phylum level), Campylobacterota was only present in the stomach of the CeD group. A comparison of the abundance of bacteria between the stomach and duodenum showed significant differences in 10 OTUs (operational taxonomic units) in the control and 9 OTUs in the CeD group at L6 (genus) and in 8 OTUs and in 6 OTUs, respectively, at L7 (species). A significant correlation was observed between the genus Novosphingobium in stomach of CeD group and possession of the DQ2.5 and DQ 8 allele, and in the duodenum – between the DQ 8 allele and the species Blautia wexlerae. Significant differences in selected, little-known genera of bacteria suggest their potential role as new biomarkers in the development of CeD. To fully understand the mechanism of CeD development in genetically predisposed individuals, it is necessary to take into account not only the abundance of a given genus or species of bacteria, but also the anatomical location of its occurrence.
1 Introduction
Although genetic predisposition and exposure to gluten are considered essential triggers for the development of coeliac disease (CeD), other factors leading to a loss of immune tolerance to gluten in predisposed individuals are still under investigation. The role of the digestive tract microbiota in the development and progression of this disease is currently a very popular concept and the focus of numerous studies (Catassi et al., 2022; Leonard and Fasano, 2016; Palmieri et al., 2022; Valitutti et al., 2019; Verdu and Schuppan, 2021). Recent research findings suggest that changes in the microbiome could precede the development of CeD or accompany the development of inflammation (Catassi et al., 2022; Valitutti et al., 2019) Increasingly, it is postulated in the literature that such research should be developed in order to improve and personalise the methods of prevention and treatment, as well as to monitor the course of the disease, with the use of potential new diagnostic bacterial biomarkers (Fasano et al., 2023; Verdu and Schuppan, 2021). Using samples from various body environments (saliva, pharynx, duodenum, and stool), Arcila-Galvis et al. (1922) analysed 9 selected studies to prepare a comprehensive map of microbial biomarkers along the digestive tract for CeD patients. The composition of the microbiota was assessed on the basis of next-generation sequencing methods (NGS), primarily focusing on the V3-V4 region of 16S rRNA) (Arcila-Galvis et al., 2022). While other studies have analysed the microbiota profile of the oral cavity, duodenum or large intestine (De Angelis et al., 2016; Leonard et al., 2021; Valitutti et al., 2019) none have provided results regarding the composition of the gastric microbiota in patients with CeD.
The aim of our study was to comparatively evaluate the composition of the bacterial microbiota of tissues collected simultaneously from the stomach (which to our knowledge has never been explored before) and duodenum in children with newly diagnosed CeD and the control group. We also attempted to analyse the correlation between the obtained bacterial profiles, biochemical and immunological results, clinical presentation, histopathological evaluation of duodenal biopsies, and HLA II DQ2 /DQ8 allele typing in blood samples.
2 Materials and methods
Patients
Between 2019 and 2022, the study recruited children and adolescents aged 2 to 18 years from the Department of Paediatrics, Gastroenterology and Nutrition, University Children’s Hospital in Krakow, Poland. Participants who underwent gastroscopy were divided into two groups. The first group consisted of 40 patients with newly diagnosed CeD according to the diagnostic criteria of the European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) published in 2012 (Husby et al., 2012). The second group (control) comprised 20 children in whom CeD and other gastroenterological diseases were excluded.
The exclusion criteria for the study included individuals below 2 or over 18 years of age, recent treatment with antibiotics and/or probiotics within 30 days before tissue sample collection (Elvers et al., 2020; Sroka-Oleksiak et al., 2020; Yassour et al., 2016), confirmed acute or chronic infections and inflammatory diseases in the gastrointestinal tract (especially Helicobacter pylori infection and inflammatory bowel disease), obesity, thyroid disorders, diabetes, active neoplastic diseases (especially gastrointestinal), congenital and/or acquired immune deficiencies (especially selective IgA deficiency), and lack of consent to participate in the study.
All procedures performed in this study involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments and with the ethical standards of the Jagiellonian University Bioethics Committee in Krakow, Poland (decision No. 1072.6120.82.2018). Informed consent was obtained from patients’ parents or legal guardians (for all patients under 18 years of age) and, additionally, from patients themselves if they were above 16 years old.
Samples
The children underwent routine laboratory blood testing, including fasting blood glucose, total cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), triglycerides (TGs), iron (Fe) levels and alanine aminotransferase (ALT). Tissue transglutaminase IgA antibodies (anti-tTG) were assessed in all enrolled children using an enzyme-linked immunosorbent assay (ELISA), with levels greater than 20 RU/ml considered abnormal. Moreover, coeliac HLA DQ2/DQ8 allele typing was performed (CeliacStrip; Operon; Cuarte de Huerva, Spain). All laboratory tests were carried out at the laboratory of the University Children’s Hospital in Krakow, in accordance with the principles of Good Laboratory Practice (GLP) and using tests certified by In Vitro Diagnostics/Food and Drug Administration (IVD/FDA) intended for routine medical diagnostics and included in the clinical protocol of specialised laboratories at this hospital. Double biopsy samples were taken from stomach and duodenum during gastroscopy. One pair of biopsy specimens was evaluated histopathologically to exclude inflammation and villous atrophy in the control group and to assess the severity of lesions in patients with CeD, according to the Marsh-Oberhuber classification (Oberhuber, 2000). The other pair was immediately frozen at −80 °C but for no longer than 2 months and transported under deep-freeze conditions to the Department of Molecular Medical Microbiology, Division of Microbiology, Jagiellonian University Medical College in Krakow. Subsequently, library preparation and sequencing were performed for microbiome analysis.
Next generation sequencing
To obtain high-quality sequence reads from low biomass samples, such as the stomach and duodenum, we followed a protocol described by Sroka-Oleksiak et al. (2020), i.e. we used the Nested-PCR method, which requires two primer sets and two successive PCR reactions. This procedure enabled to increase the specificity and sensitivity of the isolates amplified for sequencing (Sroka-Oleksiak et al., 2020).
Microbiological data obtained at 6 taxonomic levels (phylum [L2], class [L3], order [L4], family [L5], genus [L6] and species [L7]) were compared between groups (stomach of CeD vs stomach of control group and duodenum of CeD vs duodenum of control group). We also conducted a comparison of samples within the CeD group (stomach vs duodenum) and within the control group (stomach vs duodenum). The data obtained at the L6 and L7 were correlated with the clinical and laboratory parameters of children with CeD (glycemia, total cholesterol, LDL, HDL, TGs, ALT, creatinine, Fe levels, tTG antibodies titre, coeliac HLA DQ2/DQ8 genes, histopathological results), as well as age and body mass index (BMI).
Bioinformatic and statistical analysis
Raw sequencing reads were included in the QIIME2 analysis pipeline (Bolyen et al., 2019). In this pipeline, initial read quality control, filtering, denoising and feature table generation were performed using DADA2 software (Callahan et al., 2016). Subsequently, a phylogenetic tree was generated from the masked alignment using MAFFT software (Katoh and Toh, 2010) and FastTree software (Price et al., 2009). Alpha and beta diversity analyses were conducted through the q2-diversity plugin with 2,000 subsampled reads based on the rarefaction curve. Kruskal-Wallis or PERMANOVA tests were used to compare alpha and beta diversity parameters among groups. Taxonomic classification of reads was done using a pre-trained Naive Bayes classifier (sklearn) and the q2-feature-classifier plugin. This classifier was trained on the Weighted Silva 138 99% OTUs full-length sequences database. Finally, differential abundance analysis was conducted using MicrobiomeAnalyst software (Dhariwal et al., 2017) using edgeR approach (Robinson et al., 2009) with the Benjamini-Hochberg (false discovery rate-FDR) correction. Taxa were considered significantly altered if they had an FDR < 0.05.
Statistical analysis was carried out with IBM SPSS Statistics 28 (Armonk, NY, USA). Since the variables reflecting biochemical parameters and BMI did not consistently follow the normal distribution, comparisons between study groups were carried out using the non-parametric Mann-Whitney U test. Differences in gender and the prevalence of HLA DQ2/DQ8 alleles between the coeliac disease and control groups were assessed with Fisher’s exact test.
The dependence between the numbers of specific bacteria and quantitative clinical traits was evaluated using correlation analysis, in which traits deviating from the normal distribution were analysed using Spearman’s rank correlation coefficient. A p-value of less than 0.05 was considered significant. The association between DQ gene alleles and selected microbiota was tested using Student’s t-test with the JASP (https://jasp-stats.org/) statistical software.
3 Results
Characteristics of the study population
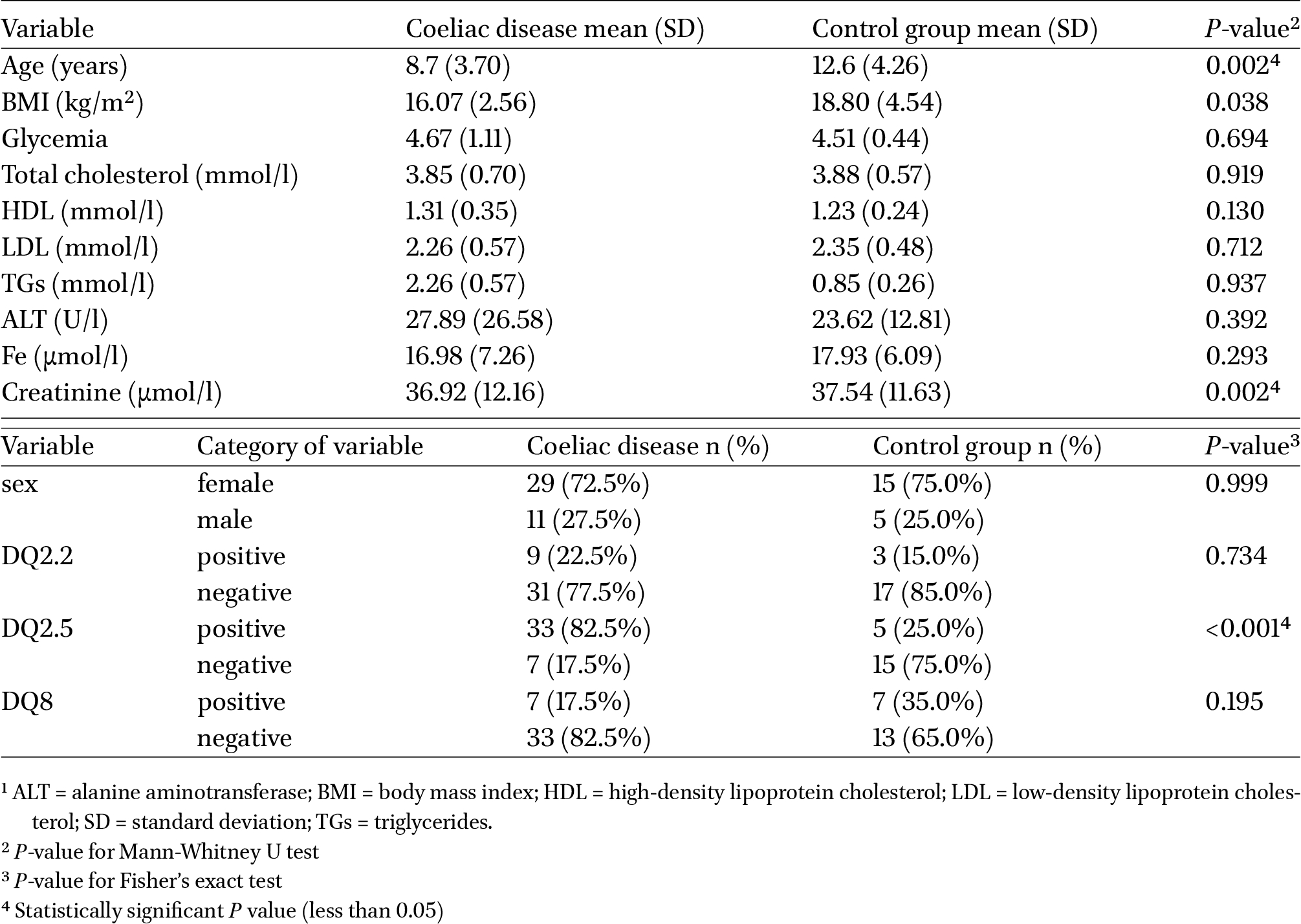
Clinical data concerning both the studied group of patients and the control group are presented in Table 1. The analysis revealed statistically significant differences between the CeD and control groups in terms of age, creatinine levels and the carriage of the HLA DQ 2.5 allele.

Clinical characteristics of the study groups1
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00009
Based on histopathological examination of duodenal biopsies, it was found that, in the CeD group, 5 children met the diagnostic criteria for type 3a, 11 children for type 3b, 23 children for 3c and one patient for type 2 villous atrophy according to the Oberhuber-Marshal classification (Oberhuber, 2000). The mean number of intraepithelial lymphocytes per 100 enterocytes (IEL) was 56.85 (±23.91; ranging from 28 IEL to 100 IEL).
Metagenomic sequencing
Sequencing was performed on 60 tissue samples as follows:
- ∙ Stomach samples yielded a total of 4,029,168 paired reads, 67,152.8 reads on average per sample. The number of reads per sample ranged from a minimum of 19,906 to a maximum of 150,821, with a median of 60,110.0.
- ∙ Duodenum samples generated 4,435,481 paired reads, 75,177.644068 reads on average per sample. The number of reads per sample ranged from a minimum of 17,277 to a maximum of 780,440, with a median of 58,465.0.
The DNA sequences obtained from gastric and duodenal biopsies corresponded to a total of, respectively, 62 and 63 OTUs at the genus level (L6). At the species level (L7), 50 and 46 OTUs were detected, respectively. The reads were clustered during preliminary filtering for 97% identity, which is equivalent to sequence similarity at a given taxonomic level. Therefore, due to the possibility of errors in data interpretation, because the percentage of classified reads was below 97% at the species level (L7), the microbiota composition at L7 and correlation analyses were conducted for selected clinical data and the numbers of only those OTUs which were clearly assigned to a specific species.
Biodiversity
The alpha diversity profiling showed no statistical significance in sample diversity between CeD patients and controls from either the stomach or duodenum. Similarly, when comparing the diversity between the stomach and duodenum within each group, the results showed no significant variations (Table 2).
The beta diversity analysis revealed no significant differences between the gastric and duodenal samples in both groups (P > 0.05). However, significant dissimilarities were observed in the gastric samples when comparing the CeD group to the control (Bray-Curtis index, P = 0.008, and weighted UniFrac distance, P = 0.024) and borderline significance in the duodenal samples (Jaccard index, P = 0.049; Table 2).
Alpha and beta diversity using the most common metrics in tissue samples from paediatric patients with CeD and a control group of children
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00009
Abundance
We conducted a systematic assessment of the bacterial profile of the tissue samples at the L2 taxonomic level to provide an overall picture. For a more detailed analysis of the differences in the composition of the gastric and duodenal microbiota, evaluations at the L6 and L7 taxonomic levels were carried out.
Due to the large number of taxa at L6 (62 in the stomach, 63 in the duodenum) and L7 (50 in the stomach, 46 in the duodenum) and the fact that 97% of reads were classified at the species level, the description in this article refers only to a few selected, statistically significant observations. Taxa with percentages lower than 1% and/or no significant differences between the compared groups were collectively categorised as ‘other’.
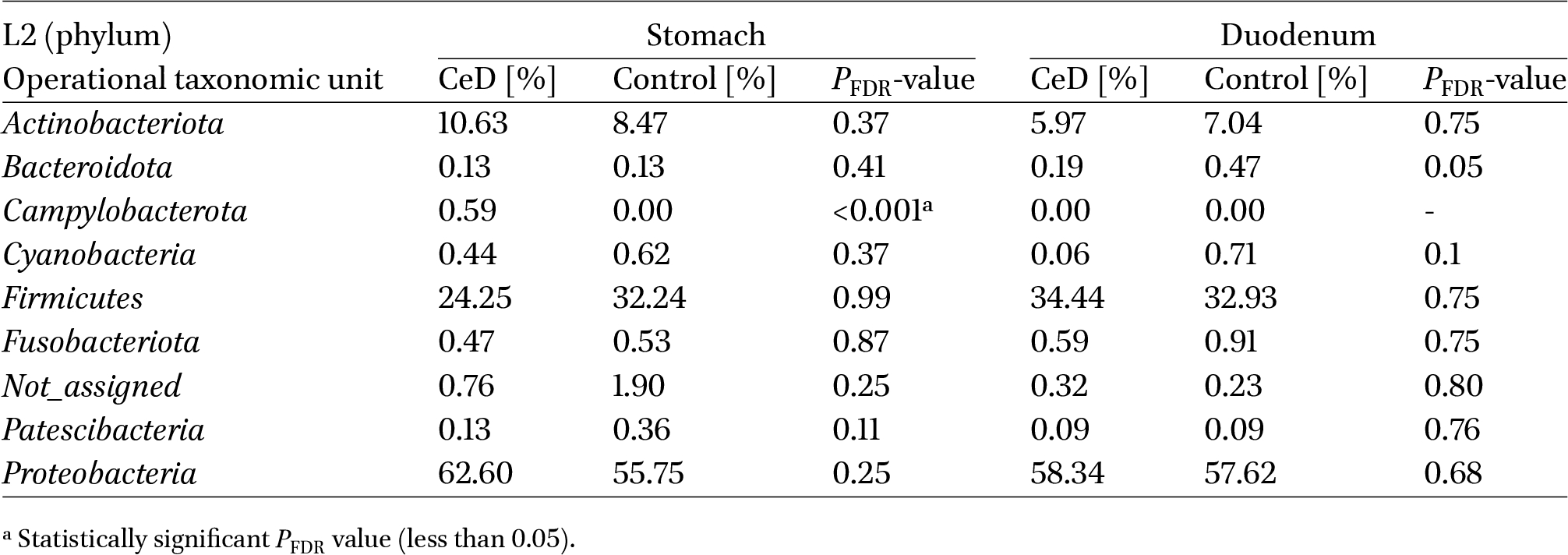
Proteobacteria were the dominant phylum in both study groups in the stomach and duodenum. The phyla Firmicutes and Actinobacteriota were also prominent (Table 3). We found no significant differences as to the relative percentages of most bacterial phyla between groups and anatomical locations, except for the phylum Campylobacterota, which was only present in the stomach of children with CeD.
Comparison of the relative percentage of bacteria at the phylum level (L2) between groups: CeD and control, in the stomach and in the duodenum
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00009
At the L6 level, both in the stomach and duodenum, 11 OTUs were identified which corresponded to bacterial genera and exhibited statistically significant differences in relative percentages between the patient groups (Supplementary Table S1). A comparison of the abundance of individual bacterial genera between the stomach and duodenum showed significant differences in 10 OTUs in the control group and 9 OTUs in the group of children with CeD (Supplementary Table S1).
At the L7 taxonomic level, we identified 8 OTUs in the stomach and 9 OTUs in the duodenum, corresponding to bacterial species with statistically significant differences in relative percentages between the patient groups (Supplementary Table S2). A comparison of the abundance of individual bacterial species between the stomach and duodenum showed significant differences in 8 OTUs in the control group and 6 OTUs in the group of CeD patients (Supplementary Table S2).
Correlation analysis
In the stomach but also in the duodenum, positive and negative correlations with selected laboratory and clinical data were observed at L6 and L7 levels between some taxa and: BMI, total cholesterol, LDL, HDL, TGs, Fe levels, ALT and anti-tTG titre (Supplementary Tables S3 and S4). Additionally, at L6, bacteria (in biopsies from the stomach and duodenum) exhibited correlations with creatinine concentration and (in the duodenal samples) with IEL (Supplementary Table S3). At L7, a relationship was observed between selected taxa and creatinine level, specifically in the duodenum (Supplementary Table S4).
Analysis of HLA DQ2/DQ8 allele occurrence in relation to gastric bacterial profiles in the CeD group showed a significant correlation only with bacteria belonging to the genus Novosphingobium: possession of the DQ 2.5 allele resulted in lower abundance, while possession of the DQ 8 allele led to a greater abundance of these bacteria (Figure 1). In the duodenum, a positive association was observed between the presence of the DQ 8 allele and the abundance of bacteria belonging to the genus Blautia, but a statistically significant relationship was observed only for the species Blautia wexlerae (Figure 1). No statistically significant correlations were observed for the remaining parameters.
Significant relationship between the occurrence of specific HLA DQ alleles in children with coeliac disease and bacterial abundance in: (A) stomach, (B) duodenum.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00009
4 Discussion
Data from many studies indicate that particular changes in the composition of the gut microbiota may precede the onset of CeD in genetically susceptible individuals. The alterations in the microbiota profile and resulting changes in specific metabolic pathways could potentially lead to a shift from gluten tolerance to an immune response to gluten (Leonard and Fasano, 2016). In our study, we focused on assessing the composition of the tissue microbiota, not only in the duodenal but also in the gastric mucus of young patients with newly diagnosed CeD which, to the best of our knowledge, has never been done before. This study was based on the analysis of the genetic material of microorganisms using NGS.
Biodiversity analysis did not show statistically significant differences concerning alpha diversity among the studied samples (Table 2). Similar results were obtained in three other studies (Federica et al., 2022; Leonard et al., 2021; Palmieri et al., 2022). In contrast, a comparison of beta diversity between the two studied groups seems to indicate a significant difference (but only in single metrics) in the number and abundance of bacteria in gastric samples (Bray-Curtis index) and duodenal samples (Jaccard index) between CeD patients and the control group. The phylogenetic relationship (weighted UniFrac distance) between the microorganisms found in the gastric samples of the two study groups was also statistically significant (Table 2), indicating a low phylogenetic affinity between these groups.
Comparison of bacterial profiles at the highest taxonomic level (phylum – L2) revealed the presence of the Campylobacterota phylum only in patients with CeD (in the stomach) (Table 3). Campylobacterota, previously known as Epsilonproteobacteria, is now classified as a phylum consisting of a large group of mostly Gram-negative, spiral-shaped motile bacteria, including Sulfurospirillum spp., Helicobacter spp., and Campylobacter spp., among others. H. pylori is the most well-known and the most prevalent Helicobacter species in the human stomach (Matos et al., 2021). Although H. pylori remains one of the bacterial species that is most likely to trigger autoimmunity, various studies have either failed to define its causal role, as Yue et al. (2022) indicated in their meta-analysis, or did not show a significant association between H. pylori and CeD, as shown by the cross-sectional study by Basyigit et al. (2017). Our study involved children in whom the presence of H. pylori colonisation was excluded, and the sequencing results did not indicate the presence of its genetic material in the samples. In contrast, at L6, we found a significant relative percentage of bacteria belonging to the Helicobacter genus (significantly higher in the stomach than in the duodenum) in patients with CeD but not in the control group. Presumably, this is a group of other gastric, spiral-shaped non-Helicobacter pylori helicobacters (NHPH) which are associated with gastrointestinal disorders. Some of the known NHPH species, such as Helicobacter suis, Helicobacter felis, Helicobacter bizzozeronii, Helicobacter salomonis and Helicobacter heilmannii, have been detected in the human stomach and may be associated with antral erosions, gastritis, duodenal ulcers, and even gastric MALT lymphoma (Matos et al., 2021; Taillieu et al., 2023). While we were unable to identify any of these specific species, the presence of the Helicobacter genus in the stomach of patients with CeD, but not in the control group, could be considered as one of the candidates for a microbial biomarker triggering CeD.
At the genus level (L6), we identified several other significant differences in bacterial abundance between the CeD and control groups. Particularly noteworthy is the genus Novosphingobium, which was exclusively found in the stomach of the CeD group and was absent in the control (Supplementary Table S1). As in the case of Helicobacter, bacteria belonging to the genus Novosphingobium were present in patients with CeD, especially in the stomach, while in the control group, it was absent. Although it was not possible to identify bacterial species of this genus in this study, at the L6, correlations with clinical data showed a significant negative (rho −0.31, P = 0.047) relationship between Novosphingobium and HDL in the stomach (Supplementary Table S3) and anti-tTG (rho −0.324, P = 0.042) in the duodenum, and a positive correlation with creatinine (rho 0.34, P = 0.034) in the duodenum of patients with CeD (Supplementary Table S4). We observed an intriguing significant relationship between the genus Novosphingobium and the presence of selected HLA DQ genes: a negative correlation with DQ2.5 (P = 0.029, Mann-Whitney U test) and a positive correlation with DQ8 (P = 0.034, Mann-Whitney U test) (Figure 1). The genus Novosphingobium, classified in the class Alphaproteobacteria and family Erythrobacteraceae, comprises Gram-negative, rod-shaped non-spore-forming bacteria widely distributed in various environments, such as toxic chemical-contaminated soil and fresh and sea water (Y. Liu et al., 2021). However, some species may be associated with the development of autoimmune diseases like primary biliary cirrhosis (PBC). In a review about alphaproteobacteria causing such diseases, Mohammed and Mattner cited reports associating Novosphingobium aromaticivorans infection with PBC. The authors proposed molecular mimicry between these bacteria and mammalian proteins as a possible mechanism. This relationship between mammalian and bacterial structures can trigger immunological cross-reactions leading to tissue pathology and autoimmunity (Mohammed and Mattner, 2009). Moreover, PBC frequently coexists with CeD (Kingham and Parker, 1998). A similar mechanism has also been described by Galipeau and Verdu in their analysis, citing the study of Petersen et al. who demonstrated that DQ2.5-restricted gliadin epitopes shared high similarity with peptides from Pseudomonas fluorescens and Pseudomonas aeruginosa. They postulated that cross-reactivity could occur between other bacterial or viral peptides and gliadin, potentially initiating or maintaining CeD (Galipeau and Verdu, 2022). Thus, it is also possible that both bacteria belonging to the genus Novosphingobium and other microorganisms exert similar effects on the development of autoimmune diseases in predisposed individuals, including CeD.
In our study, we observed a noteworthy relationship between low HDL and high creatinine levels, the occurrence of DQ8 allele, and the percentage of bacteria of the genus Novosphingobium in the stomach of CeD children. These findings suggest that these microorganisms could be involved in the disruption of homeostasis in humans, and it is possible that a specific species within this genus, distinct from N. aromaticivorans, could play a role in the development of CeD. Interestingly, the correlation between the abundance of Novosphingobium genus in the duodenum and the level of anti-tTG antibodies is negative, suggesting a potentially beneficial effect of these microorganisms on the disease course in children with CeD. Such a hypothesis aligns with reports presented in the research review by Caputo et al. (2010), in which one of the possible treatment strategies, instead of gluten free diet (GFD), involves using prolyl oligopeptides (large cytosolic enzymes that belong to the class of serine peptidases) to eliminate harmful gluten peptides from the coeliac diet, thereby neutralising the immunogenic effect of the gluten epitopes. Notably, such enzymes are produced by, among others, Novosphingobium capsulatum (homotypic synonym: Sphingomonas capsulata) (Caputo et al., 2010).
We also observed a significant negative correlation (rho-0.034, P = 0.034) between IEL in the duodenum and the relative percentage of bacteria belonging to the genus Blautia (Supplementary Table S4). The abundance of these microorganisms, particularly the species B. wexlerae, was significantly higher in the duodenum of CeD children compared to the control group, and interestingly, these microorganisms were also more abundant in the duodenum than in the stomach of the same patients (Supplementary Tables S1 and S2). In addition, the relative percentage of Blautia positively correlated with the presence of the HLA DQ 8 gene, as in the case of Novosphingobium bacteria. Specifically, a statistically significant relationship (P = 0.011, Mann-Whitney U test) was found between the HLA DQ 8 gene and the species B. wexlerae (Figure 1). Some Clostridium and Ruminococcus species have been reclassified as Blautia based on phenotypic and phylogenetic analyses. Blautia is a genus that comprises obligately anaerobic, nonsporulating, coccobacillus-shaped Gram-positive bacteria, commonly found in the feces and intestines of mammals. Within this genus, B. wexlerae and B. luti have been identified as the most abundant species, especially in the human intestine. Although these bacteria are believed to possess probiotic properties, their role in health and disease has not yet been fully explained (X. Liu et al., 2021). Blautia members are associated with the production of short-chain fatty acids (SCFAs) and antimicrobial peptides. Additionally, these bacteria have been found to be decreased in the microbiota of individuals with obesity or diabetes (Benı́tez-Páez et al., 2020a; Maturana and Cárdenas, 2021). Studies analysing the composition of the intestinal microbiota in people with CeD have yielded various results. Shi T. et al., in their examination of faecal samples from adults with newly diagnosed CeD, reported a significantly lower abundance of bacteria from the genus Blautia compared to the control group (Shi et al., 2022). Similarly, Palmieri et al. observed a reduction in Blautia bacteria abundance in faecal samples of coeliac patients who were in remission and had followed a GFD for at least 2 years, when compared to the control group (Palmieri et al., 2022). Additionally, Sample et al. analysed stool samples from children with CeD and found a unique microbial signature in these patients when placed on a GFD for one year, including significantly lower abundance of Blautia bacteria, compared to the control group (Sample et al., 2021). Moreover, according to Hansen et al., a low-gluten diet resulted in a decrease in B. wexlerae abundance compared to a high-gluten diet (Hansen et al., 2018). In our study, we observed a significantly higher relative percentage of bacteria from the genus Blautia in the duodenum of patients with CeD compared to children from the control group. This finding can be attributed to two factors. Firstly, our assessment focused on the microbiota of tissues from the upper digestive tract rather than faecal samples. Secondly, the CeD group in our study consisted of newly diagnosed individuals who had not yet excluded gluten from their diet, which may explain the increased abundance of this bacterium species. On the other hand, a negative correlation with IEL suggests a potential anti-inflammatory effect of these bacteria. It seems to confirm the supposition of other researchers regarding the role of the genus Blautia, and particularly the species B. wexlerae, in fat metabolism, obesity reduction and anti-inflammatory effects (Benı́tez-Páez et al., 2020; Hosomi et al., 2022).
The negative correlation between the relative percentage of bacteria from the Novosphingobium genus and the presence of the HLA DQ2.5 gene, as well as the positive relationship between these microorganisms and bacteria from the Blautia genus with the HLA DQ8 gene, may suggest that certain species or strains of the mentioned genera may exert distinct effects, beneficial or harmful, depending on the particular HLA allele possessed by coeliac patients and on the significant abundance of these bacteria in either the stomach or duodenum. To fully comprehend the underlying mechanisms and functional roles of these microbial associations, further research is warranted.
The abundance of bacteria from the Turicibacter genus showed significant differences in our study, with higher levels observed in the duodenum (P < 0.001) and lower levels in the stomach compared to the duodenum (P < 0.01) in CeD group, as compared to the control group (Supplementary Table S1). Nevertheless, a significant (negative) correlation between this genus and total cholesterol (rho −0.36, P = 0.022) and LDL (rho −0.40, P = 0.01) concentrations was found in the stomach (Supplementary Table S3). Turicibacter belongs to the Firmicutes phylum and is classified as Gram-positive, rod-shaped bacteria commonly found in the human faecal microbiota (Lynch et al., 2023a; Rahman et al., 2023). Interestingly, analyses of the intestinal microbiota done by Rodriguez-Diaz et al. (2022) showed higher abundances of Turicibacter in patients with nonalcoholic fatty liver disease (NAFLD), coexisting with significant liver fibrosis, compared to NAFLD patients without or with mild liver fibrosis (Rodriguez-Diaz et al., 2022). Furthermore, in a mouse model, Li et al. (2020) observed a significantly higher abundance of, among others, the Turicibacter bacteria in the non-alcoholic steatohepatitis (NASH) group compared to the non-alcoholic fatty liver (NAFL) group, suggesting its potential as a distinguishing factor between the two conditions (Li et al., 2020). Previous studies on the human gut microbiota have shown that Turicibacter has the ability to modify bile acids and lipid metabolism of the host. The effect of diverse Turicibacter isolates on host metabolites, including lipids and bile acids, was presented by Lynch et al. Conflicting reports exist, with some indicating positive and others negative correlations of Turicibacter bacteria with dietary fats and host adiposity. The authors of the cited work hypothesised that the type of dependence is related to the properties of specific bacterial strains and that the presence of bile modifying genes from these isolates influences host physiology. They found that these genes are sufficient to modify host lipid and cholesterol states (Lynch et al., 2023). Based on our clinical data and correlation results, it could be hypothesised that the stomach of children with CeD may be colonised by Turicibacter strains which have a beneficial effect on lipid metabolism and adipose tissue. However, considering the scarcity of studies on the presence of these bacteria in the upper digestive tract of children with CeD and their possible impact on lipid metabolism, liver function and the course of this disease, further comprehensive research is needed. The above findings suggest that this bacterial taxon could also serve as one of the biomarkers for CeD.
5 Conclusions
Our work indicates a significant presence of microorganisms belonging to little-known genera in the upper digestive tract of children with newly diagnosed CeD. To our knowledge, this is the first analysis involving a comprehensive assessment of bacterial profiles of both gastric and duodenal biopsies obtained from children with CeD. The results concerning the composition of the gastric microbiota and the correlation of selected genera with clinical data and specific HLA DQ genes (especially DQ 8) are particularly interesting. The analysis of the data from this study suggests that in order to fully understand the mechanism of CeD development in genetically predisposed individuals, it is necessary to take into account not only the abundance of a given genus or species of bacteria, but also the anatomical location of its occurrence. The function and correlation with the HLA gene allele may depend on whether it is present in the stomach or duodenum and in what percentage. One limitation of this study was the small number of samples, particularly in the control group. Moreover, this group was not fully representative of the general population of patients, as it was selected among patients undergoing upper endoscopy for gastrointestinal ailments that were later ruled out. Furthermore, the abundance of bacteria in these ecological niches is typically much poorer than in the lower digestive tract, as noted by Constante et al. (2022) in their work. The isolated low concentration of bacterial genetic material required additional amplification procedures (nested PCR) and ultimately limited a more precise assessment of the microbiota at the species level (L7). Due to the above-mentioned circumstances, direct comparison of our results with observations from other researchers may prove challenging, especially considering that they did not test gastric samples. However, our study highlights significant differences in selected genera of bacteria, such as Helicobacter, Novosphingobium, Blautia and Turicibacter, suggesting their potential role as new biomarkers in the development of CeD in children and adolescents. This study can also serve as a basis for further, more detailed research analysing the functions of specific species and strains within these genera of microorganisms.
Corresponding author; e-mail: tomasz.gosiewski@uj.edu.pl
Supplementary material
Supplementary material is available online at: https://doi.org/10.6084/m9.figshare.25562211
Table S1. Statistically significant (
Table S2. Statistically significant (
Table S3. Correlations between clinical data and the presence of bacteria in the stomach of patients with celiac disease at the genus (L6) and species (L7) levels.
Table S4. Correlations between clinical data and the presence of bacteria in the duodenum of patients with celiac disease at the genus (L6) and species (L7) levels.
Acknowledgements
This study was funded by the National Science Centre in Poland in frame of the project: SONATA Bis7 – 2017/26/E/NZ5/00266. The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Authors’ contribution
Conceptualisation, TG, DS, KK-D, AK, MD and AG; methodology, TG,HK, MAI and YS; software, MK and AG; validation, HK and YT; analysis of data, TG, DS, KK-D, AK, MK and AG; writing-original draft preparation, DS; writing-review and editing, TG, KK-D, AK, MK and AG; visualisation, DS and AG; supervision, TG, KK-D; funding acquisition, TG. All authors have read and agreed to the published version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Data availability
All data relevant to the study are included in the article or uploaded as supplementary information. Fastq files are available: https://portalwiedzy.cm-uj.krakow.pl/info/researchdata/UJCM77a8979a493e4aacbdceefa5121abbff/
References
Arcila-Galvis, J.E., Loria-Kohen, V., Ramı́rez de Molina, A., Carrillo de Santa Pau, E. and Marcos-Zambrano, L.J., 2022. A comprehensive map of microbial biomarkers along the gastrointestinal tract for celiac disease patients. Frontiers in Microbiology 13: 956119. https://doi.org/10.3389/fmicb.2022.956119
Basyigit, S., Unsal, O., Uzman, M., Sapmaz, F., Dogan, O.C., Kefeli, A., Asilturk, Z., Yeniova, A.O. and Nazligul, Y., 2017. Relationship between Helicobacter pylori infection and celiac disease: a cross-sectional study and a brief review of the literature. Turkey Gastroenterology Review 12: 49-54. https://doi.org/10.5114/pg.2017.65681
Benı́tez-Páez, A., Gómez del Pugar, E.M., López-Almela, I., Moya-Pérez, Á., Codoñer-Franch, P. and Sanz, Y., 2020. Depletion of Blautia species in the microbiota of obese children relates to intestinal inflammation and metabolic phenotype worsening. MSystems 5: e00857-19. https://doi.org/10.1128/msystems.00857-19
Bolyen, E., Rideout, J.R., Dillon, M.R., Bokulich, N.A., Abnet, C.C., Al-Ghalith, G.A., Alexander, H., Alm, E.J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J.E., Bittinger, K., Brejnrod, A., Brislawn, C.J., Brown, C.T., Callahan, B.J., Caraballo-Rodrı́guez, A.M., Chase, J., Cope, E.K., Da Silva, R., Diener, C., Dorrestein, P.C., Douglas, G.M., Durall, D.M., Duvallet, C., Edwardson, C.F., Ernst, M., Estaki, M., Fouquier, J., Gauglitz, J.M., Gibbons, S.M., Gibson, D.L., Gonzalez, A., Gorlick, K., Guo, J., Hillmann, B., Holmes, S., Holste, H., Huttenhower, C., Huttley, G.A., Janssen, S., Jarmusch, A.K., Jiang, L., Kaehler, B.D., Kang, K.B., Keefe, C.R., Keim, P., Kelley, S.T., Knights, D., Koester, I., Kosciolek, T., Kreps, J., Langille, M.G.I., Lee, J., Ley, R., Liu, Y.X., Loftfield, E., Lozupone, C., Maher, M., Marotz, C., Martin, B.D., McDonald, D., McIver, L.J., Melnik, A.V., Metcalf, J.L., Morgan, S.C., Morton, J.T., Naimey, A.T., Navas-Molina, J.A., Nothias, L.F., Orchanian, S.B., Pearson, T., Peoples, S.L., Petras, D., Preuss, M.L., Pruesse, E., Rasmussen, L.B., Rivers, A., Robeson, M.S., Rosenthal, P., Segata, N., Shaffer, M., Shiffer, A., Sinha, R., Song, S.J., Spear, J.R., Swafford, A.D., Thompson, L.R., Torres, P.J., Trinh, P., Tripathi, A., Turnbaugh, P.J., Ul-Hasan, S., van der Hooft, J.J.J., Vargas, F., Vázquez-Baeza, Y., Vogtmann, E., von Hippel, M., Walters, W., Wan, Y., Wang, M., Warren, J., Weber, K.C., Williamson, C.H.D., Willis, A.D., Xu, Z.Z., Zaneveld, J.R., Zhang, Y., Zhu, Q., Knight, R. and Caporaso, J.G., 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852-857. https://doi.org/10.1038/s41587-019-0209-9
Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J.A. and Holmes, S.P., 2016. DADA2: high-resolution sample inference from illumina amplicon data. Nature Methods 13: 581-583. https://doi.org/10.1038/nmeth.3869
Caputo, I., Lepretti, M., Martucciello, S. and Esposito, C., 2010. Enzymatic strategies to detoxify gluten: implications for celiac disease. Enzyme Research 2010: 174354. https://doi.org/10.4061/2010/174354
Catassi, C., Verdu, E.F., Bai, J.C. and Lionetti, E., 2022. Coeliac disease. The Lancet 399: 2413-2426. https://doi.org/10.1016/S0140-6736(22)00794-2
Constante, M., Libertucci, J., Galipeau, H.J., Szamosi, J.C., Rueda, G., Miranda, P.M., Pinto-Sanchez, M.I., Southward, C.M., Rossi, L., Fontes, M.E., Chirdo, F.G., Surette, M.G., Bercik, P., Caminero, A. and Verdu, E.F., 2022. Biogeographic variation and functional pathways of the gut microbiota in celiac disease. Gastroenterology 163: 1351-1363.e15. https://doi.org/10.1053/j.gastro.2022.06.088
De Angelis, M., Vannini, L., Di Cagno, R., Cavallo, N., Minervini, F., Francavilla, R., Ercolini, D. and Gobbetti, M., 2016. Salivary and fecal microbiota and metabolome of celiac children under gluten-free diet. International Journal of Food Microbiology 239: 125-132. https://doi.org/10.1016/j.ijfoodmicro.2016.07.025
Dhariwal, A., Chong, J., Habib, S., King, I.L., Agellon, L.B. and Xia, J., 2017. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Research 45: W180-W188. https://doi.org/10.1093/nar/gkx295
Elvers, K.T., Wilson, V.J., Hammond, A., Duncan, L., Huntley, A.L., Hay, A.D. and van der Werf, E.T., 2020. Antibiotic-induced changes in the human gut microbiota for the most commonly prescribed antibiotics in primary care in the UK: a systematic review. BMJ Open 10: e035677. https://doi.org/10.1136/bmjopen-2019-035677
Fasano, A., Leonard, M.M., Kenyon, V., Valitutti, F., Pennacchio-Harrington, R., Piemontese, P., Francavilla, R., Norsa, L., Passaro, T., Crocco, M., Baldassarre, M. and Trovato, C.M., 2023. Cohort profile: celiac disease genomic, environmental, microbiome and metabolome study; a prospective longitudinal birth cohort study of children at-risk for celiac disease. PLoS ONE 18: e282739. https://doi.org/10.1371/journal.pone.0282739
Federica, R., Edda, R., Daniela, R., Simone, B., Giulia, N., Gabriele, L., Marta, M., Marco, P., Gianluca, B., Elena, N., Matteo, C., Serena, S., Matteo, R., Amedeo, A. and Salvatore, C.A., 2022. Characterization of the ‘gut microbiota-immunity axis’ and microbial lipid metabolites in atrophic and potential celiac disease. Frontiers in Microbiology 13: 886008. https://doi.org/10.3389/fmicb.2022.886008
Galipeau, H.J. and Verdu, E.F., 2022. The double-edged sword of gut bacteria in celiac disease and implications for therapeutic potential. Mucosal Immunology 15: 235-243. https://doi.org/10.1038/s41385-021-00479-3
Hansen, L.B.S., Roager, H.M., Søndertoft, N.B., Gøbel, R.J., Kristensen, M., Vallès-Colomer, M., Vieira-Silva, S., Ibrügger, S., Lind, M.V., Mærkedahl, R.B., Bahl, M.I., Madsen, M.L., Havelund, J., Falony, G., Tetens, I., Nielsen, T., Allin, K.H., Frandsen, H.L., Hartmann, B., Holst, J.J., Sparholt, M.H., Holck, J., Blennow, A., Moll, J.M., Meyer, A.S., Hoppe, C., Poulsen, J.H., Carvalho, V., Sagnelli, D., Dalgaard, M.D., Christensen, A.F., Lydolph, M.C., Ross, A.B., Villas-Bôas, S., Brix, S., Sicheritz-Pontén, T., Buschard, K., Linneberg, A., Rumessen, J.J., Ekstrøm, C.T., Ritz, C., Kristiansen, K., Nielsen, H.B., Vestergaard, H., Færgeman, N.J., Raes, J., Frøkiær, H., Hansen, T., Lauritzen, L., Gupta, R., Licht, T.R. and Pedersen, O., 2018. A low-gluten diet induces changes in the intestinal microbiome of healthy Danish adults. Nature Communications 9: 4630. https://doi.org/10.1038/s41467-018-07019-x
Hosomi, K., Saito, M., Park, J., Murakami, H., Shibata, N., Ando, M., Nagatake, T., Konishi, K., Ohno, H., Tanisawa, K., Mohsen, A., Chen, Y.A., Kawashima, H., Natsume-Kitatani, Y., Oka, Y., Shimizu, H., Furuta, M., Tojima, Y., Sawane, K., Saika, A., Kondo, S., Yonejima, Y., Takeyama, H., Matsutani, A., Mizuguchi, K., Miyachi, M. and Kunisawa, J., 2022. Oral administration of Blautia wexlerae ameliorates obesity and type 2 diabetes via metabolic remodeling of the gut microbiota. Nature Communications 13: 4477. https://doi.org/10.1038/s41467-022-32015-7
Husby, S., Koletzko, S., Korponay-Szabó, I.R., Mearin, M.L., Phillips, A., Shamir, R., Troncone, R., Giersiepen, K., Branski, D., Catassi, C., Lelgeman, M., Mäki, M., Ribes-Koninckx, C., Ventura, A. and Zimmer, K.P., 2012. European society for pediatric gastroenterology, hepatology, and nutrition guidelines for the diagnosis of coeliac disease. Journal of Pediatric Gastroenterology and Nutrition 54: 136-160. https://doi.org/10.1097/MPG.0b013e31821a23d0
Katoh, K. and Toh, H., 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. https://doi.org/10.1093/bioinformatics/btq224
Kingham, J.G.C. and Parker, D.R., 1998. The association between primary biliary cirrhosis and coeliac disease: a study of relative prevalences. Gut 42: 120-122. https://doi.org/10.1136/gut.42.1.120
Leonard, M.M. and Fasano, A., 2016. The microbiome as a possible target to prevent celiac disease. Expert Review of Gastroenterology and Hepatology 10: 555-556. https://doi.org/10.1586/17474124.2016.1166954
Leonard, M.M., Valitutti, F., Karathia, H., Pujolassos, M., Kenyon, V., Fanelli, B., Troisi, J., Subramanian, P., Camhi, S., Colucci, A., Serena, G., Cucchiara, S., Trovato, C.M., Malamisura, B., Francavilla, R., Elli, L., Hasan, N.A., Zomorrodi, A.R., Colwell, R. and Fasano, A., 2021. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proceedings of the National Academy of Sciences of the USA 118: e2020322118. https://doi.org/10.1073/pnas.2020322118
Li, C., Cui, L., Wang, X., Yan, Z., Wang, S. and Zheng, Y., 2020. Using intestinal flora to distinguish non-alcoholic steatohepatitis from non-alcoholic fatty liver. Journal of International Medical Research 48. https://doi.org/10.1177/0300060520978122
Liu, X., Mao, B., Gu, J., Wu, J., Cui, S., Wang, G., Zhao, J., Zhang, H. and Chen, W., 2021. Blautia – a new functional genus with potential probiotic properties? Gut Microbes 13: 1-21, 1875796, https://doi.org/10.1080/19490976.2021.1875796
Liu, Y., Pei, T., Du, J., Huang, H., Deng, M.R. and Zhu, H., 2021. Comparative genomic analysis of the genus Novosphingobium and the description of two novel species Novosphingobium aerophilum sp. nov. and Novosphingobium jiangmenense sp. nov. Systematic and Applied Microbiology 44: 126202. https://doi.org/10.1016/j.syapm.2021.126202
Lynch, J.B., Gonzalez, E.L., Choy, K., Faull, K.F., Jewell, T., Arellano, A., Liang, J., Yu, K.B., Paramo, J. and Hsiao, E.Y., 2023. Gut microbiota Turicibacter strains differentially modify bile acids and host lipids. Nature Communications 14: 3669. https://doi.org/10.1038/s41467-023-39403-7
Matos, R., Amorim, I., Magalhães, A., Haesebrouck, F., Gärtner, F. and Reis, C.A., 2021. Adhesion of Helicobacter species to the human gastric mucosa: a deep look into glycans role. Frontiers in Molecular Biosciences 8: 656439. https://doi.org/10.3389/fmolb.2021.656439
Maturana, J.L. and Cárdenas, J.P., 2021. Insights on the evolutionary genomics of the Blautia genus: potential new species and genetic content among lineages. Frontiers in Microbiology 12: 660920. https://doi.org/10.3389/fmicb.2021.660920
Mohammed, J.P. and Mattner, J., 2009. Autoimmune disease triggered by infection with alphaproteobacteria. Expert Review of Clinical Immunology. https://doi.org/10.1586/eci.09.23
Oberhuber, G., 2000. Histopathology of celiac disease. Biomedicine and Pharmacotherapy 54: 368-372. https://doi.org/10.1016/S0753-3322(01)80003-2
Palmieri, O., Castellana, S., Bevilacqua, A., Latiano, A., Latiano, T., Panza, A., Fontana, R., Ippolito, A.M., Biscaglia, G., Gentile, A., Gioffreda, D., Decina, I., Tricarico, M., Sinigaglia, M., Corbo, M.R., Mazza, T., Perri, F. and Lamacchia, C., 2022. Adherence to gluten-free diet restores alpha diversity in celiac people but the microbiome composition is different to healthy people. Nutrients 14: 2452. https://doi.org/10.3390/nu14122452
Price, M.N., Dehal, P.S. and Arkin, A.P., 2009. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution 26: 1641-1650. https://doi.org/10.1093/molbev/msp077
Rahman, A.T., Shin, J., Whang, C.H., Jung, W., Yoo, D., Seo, C., Cho, B.K. and Jon, S., 2023. Bilirubin nanomedicine rescues intestinal barrier destruction and restores mucosal immunity in colitis. ACS Nano 17: 10996-11013. https://doi.org/10.1021/acsnano.3c03252
Robinson, M.D., McCarthy, D.J. and Smyth, G.K., 2009. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139-140. https://doi.org/10.1093/bioinformatics/btp616
Rodriguez-Diaz, C., Taminiau, B., Garcı́a-Garcı́a, A., Cueto, A., Robles-Dı́az, M., Ortega-Alonso, A., Martı́n-Reyes, F., Daube, G., Sanabria-Cabrera, J., Jimenez-Perez, M., Isabel Lucena, M., Andrade, R.J., Garcı́a-Fuentes, E. and Garcı́a-Cortes, M., 2022. Microbiota diversity in nonalcoholic fatty liver disease and in drug-induced liver injury. Pharmacological Research 182: 106348. https://doi.org/10.1016/j.phrs.2022.106348
Sample, D., Fouhse, J., King, S., Huynh, H.Q., Dieleman, L.A., Willing, B.P. and Turner, J., 2021. Baseline fecal microbiota in pediatric patients with celiac disease is similar to controls but dissimilar after 1 year on the gluten-free diet. JPGN Reports 2: e127. https://doi.org/10.1097/pg9.0000000000000127
Shi, T., Feng, Y., Liu, W., Liu, H., Li, T., Wang, M., Li, Z., Lu, J., Abudurexiti, A., Maimaitireyimu, A., Hu, J. and Gao, F., 2022. Characteristics of gut microbiota and fecal metabolomes in patients with celiac disease in Northwest China. Frontiers in Microbiology 13: 1020977. https://doi.org/10.3389/fmicb.2022.1020977
Sroka-Oleksiak, A., Młodzińska, A., Bulanda, M., Salamon, D., Major, P., Stanek, M. and Gosiewski, T., 2020. Metagenomic analysis of duodenal microbiota reveals a potential biomarker of dysbiosis in the course of obesity and type 2 diabetes: a pilot study. Journal of Clinical Medicine 9: 369. https://doi.org/10.3390/jcm9020369
Taillieu, E., De Witte, C., De Schepper, H., Van Moerkercke, W., Rutten, S., Michiels, S., Arnst, Y., De Bruyckere, S., Francque, S., van Aert, F., George, C., Callewaert, E., Callewaert, T., Vanneste, G., Vanderstraeten, E., Van Heddegem, N., Vansteelant, M., Chiers, K., Haesebrouck, F. and Van Steenkiste, C., 2023. Clinical significance and impact of gastric non-Helicobacter pylori Helicobacter species in gastric disease. Alimentary Pharmacology and Therapeutics 57: 1432-1444. https://doi.org/10.1111/apt.17488
Valitutti, F., Cucchiara, S. and Fasano, A., 2019. Celiac disease and the microbiome. Nutrients 11: 2403. https://doi.org/10.3390/nu11102403
Verdu, E.F. and Schuppan, D., 2021. Co-factors, microbes, and immunogenetics in celiac disease to guide novel approaches for diagnosis and treatment. Gastroenterology 161: 1395-1411.e4. https://doi.org/10.1053/j.gastro.2021.08.016
Yassour, M., Vatanen, T., Siljander, H., Hämäläinen, A.M., Härkönen, T., Ryhänen, S.J., Franzosa, E.A., Vlamakis, H., Huttenhower, C., Gevers, D., Lander, E.S., Knip, M. and Xavier, R.J., 2016. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Science Translational Medicine 8: 343-381. https://doi.org/10.1126/scitranslmed.aad0917
Yue, M., Chen, Q., Zhou, X., Li, L. and Lu, C., 2022. Is Helicobacter pylori infection associated with celiac disease? A meta-analysis. Turkish Journal of Gastroenterology 33: 205-212. https://doi.org/10.5152/tjg.2022.21360



{kind=link}
{kind=link}
{kind=link}
{kind=link}