Abstract
COVID-19 is caused by an airborne virus, SARS-CoV-2. The upper respiratory tract (URT) is, therefore, the first system to endure the attack. Inhabited by an assemblage of microbial communities, a healthy URT wards off the invasion. However, once invaded, it becomes destabilised, which could be crucial to the establishment and progression of the infection. We examined 696 URT samples collected from 285 COVID-19 patients at three time-points throughout their hospital stay and 100 URT samples from 100 healthy controls. We used 16S ribosomal RNA sequencing to evaluate the abundance of various bacterial taxa, α-diversity, and β-diversity of the URT microbiome. Ordinary least squares regression was used to establish associations between the variables, with age, sex, and antibiotics as covariates. The URT microbiome in the COVID-19 patients was distinctively different from that of healthy controls. In COVID-19 patients, the abundance of 16 genera was significantly reduced. A total of 47 genera were specific to patients, whereas only 2 were unique to controls. The URT samples collected at admission differed more from the control than from the samples collected at later stages of treatment. The following four genera originally depleted in the patients grew significantly by the end of treatment: Fusobacterium, Haemophilus, Neisseria, and Stenotrophomonas. Our findings strongly suggest that SARS-CoV-2 caused significant changes in the URT microbiome, including the emergence of numerous atypical taxa. These findings may indicate increased instability of the URT microbiome in COVID-19 patients. In the course of the treatment, the microbial composition of the URT of COVID-19 patients tended toward that of controls. These microbial changes may be interpreted as markers of recovery.
1 Introduction
The COVID-19 pandemic has claimed 6 million lives (https://covid19.who.int/). Caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the new disease has proven to be a devastating phenomenon with an elaborate and all-pervasive infectious nature. Several studies have addressed possible mechanisms of SARS-CoV-2 infection and pathogenesis. Airborne transmission is thought to be the primary transmission route for the virus. The microenvironment of the upper respiratory tract (URT), therefore, is the first line of defence against the infection and may be crucial to its establishment and progression.
The URT is home to multiple microbial communities, or the URT microbiome, that are vital for respiratory health (Man et al., 2017). Despite a constant circulation of microbes during inhalation and exhalation, the composition of the URT microbiome remains relatively stable. However, some pathological conditions can compromise its balance. For instance, a disrupted microbiome has been reported in cases of influenza (Borges et al., 2018), respiratory syncytial virus (Grier et al., 2021), and rhinovirus (Kloepfer et al., 2017). The URT microbiome may also house opportunistic bacteria that might cause co-infections, superinfections of the lower respiratory tract, and severe complications of viral respiratory infections, including COVID-19 (Mostafa et al., 2020).
Some authors believe that, unlike other viruses, SARS-CoV-2 does not significantly impact the nasopharynx microbiome in mild COVID-19 (Braun et al., 2021; De Maio et al., 2020); more publications, however, report multidirectional and varying degrees of URT microbiome disturbances in COVID-19 patients (Engen et al., 2021; Ma et al., 2021). Current studies often report inconsistent results, which could be attributed to differences in their design, methods, statistical analysis, and subject recruitment strategies.
Despite a substantial amount of data, the exact effect of COVID-19 on the composition of the microbiome and its dynamics is still unknown. Moreover, studies rarely address changes in the URT microbiome during the antiviral therapy – a valuable measure of the outcome. Identifying and interpreting these changes could facilitate the development of effective therapies for URT dysbiosis, such as probiotics that offset the imbalance and help restore the body’s defence mechanisms.
In this study, we compared the diversity and abundance of the URT microbiome in COVID-9 patients and healthy controls to identify host-specific characteristics that could impact disease severity and outcome. We also examined short-term changes in the URT microbiome to determine which microbial shifts could be used as markers of recovery. Additionally, we addressed some limitations of previous studies and factored in several important covariates, such as patients’ medication histories, that might have been overlooked in other studies and might have affected their findings.
2 Materials and methods
Study design and participants
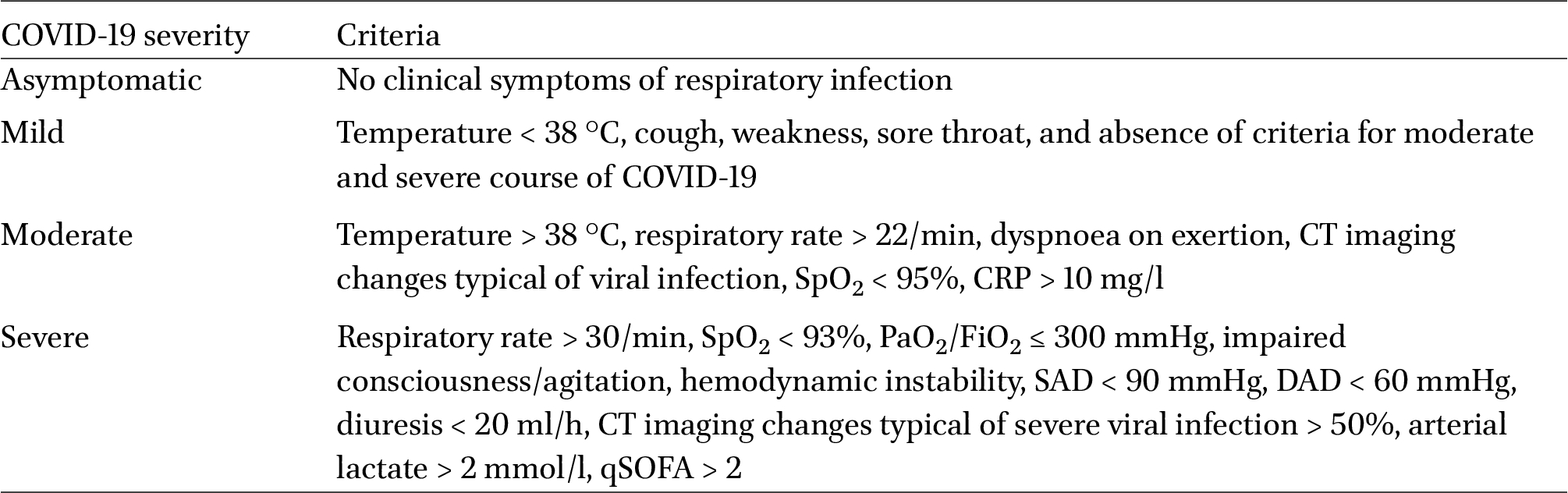
This multicentre study followed 285 SARS-CoV-2-infected patients aged 18 and above, except for cancer patients and pregnant women, over the course of their treatment at five Russian hospitals. The controls (n = 100) were above 18 years of age with no signs of acute respiratory infections or chronic disease exacerbations. Patients’ nasopharyngeal swab samples were collected upon admission to the hospital. The swabs were tested for the viral RNA using a real-time polymerase chain reaction (RT-PCR) with the AmpliSens® COVID-19-FL kit (AmpliSens®, Russia). The patients were classified by COVID-19 severity based on the following criteria: body temperature, oxygen saturation (SpO2), CT imaging changes, C-reactive protein (CRP) level, and arterial lactate level (Table 1). Two types of outcomes were registered: discharged and dead. A case report form (CRF) was filled out for every patient (Supplementary materials S1).

COVID-19 severity criteria
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Ethical considerations
The study protocol was approved by the local ethics committee of the Pediatric Research and Clinical Center for Infectious Diseases (Saint-Petersburg, Russian Federation; protocol No. 126 from June 26, 2020). All participants signed an informed consent form before enrolment.
Sample collection
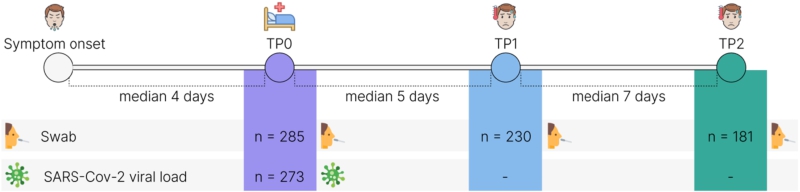
The COVID-19 patients provided 1-3 nasopharyngeal swabs for metagenomic analysis of the URT microbiome and one nasopharyngeal swab for SARS-CoV-2 viral load assessment. The healthy controls provided 1 nasopharyngeal swab for the URT microbiome analysis. The patients were instructed to gargle and rinse their mouths with regular drinking water. The posterior pharynx, sidewalls, and crypts of the tonsils were rubbed with the disposable sterile sampling swabs three to five times in a circular motion. The swabs were transported to the laboratory at 4-8 °C, frozen, and stored at −70 °C until DNA extraction. Table 2 presents the timetable for sample collection.
Timetable of swab collection from COVID-19 patients and controls
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Droplet digital PCR-based assessment of SARS-CoV-2 viral load
DNA samples were extracted using the ‘RIBO-prep’ kit (AmpliSens®, Moscow, Russia) following the manufacturer’s instructions. The precipitate was dissolved in 50 ml of the RNA buffer. Reverse transcription was performed using the ‘REVERTA-L’ kit (AmpliSens®) following the manufacturer’s instructions. cDNA samples were subsequently used in droplet digital PCR (ddPCR) carried out on the QX100 system (Bio-Rad, Hercules, CA, USA) and T100 thermal cycler (Bio-Rad). The following probe and primers (China CDC for N-gene) were used for the assessment:
- ∙ N-F GGGGAACTTCTCCTGCTAGAAT
- ∙ N-R CAGACATTTTGCTCTCAAGCTG
- ∙ N-Probe-FAM TTGCTGCTGCTTGACAGATT
The ddPCR reaction components and their volumes are shown in Supplementary Table S1. The amplification protocol is provided in Supplementary Table S2.
Results were quantified using the QX100 Droplet Reader (Bio-Rad). For cluster separation, a threshold of 5,000 relative fluorescence units (RFUs) was set for all assessments. The results were presented as the number of target cDNA copies per 20 μl.
Sequencing
DNA was extracted from 0.2-ml samples using the QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. The Illumina protocol (16S Metagenomic Sequencing Library Preparation) was used for 16S library preparation and sequencing. The DNA was amplified using standard 16S rRNA gene primers complementary to the V3-V4 region. The Phusion High-Fidelity DNA Polymerase (Thermo Fischer, Waltham, MA, USA) was used for the PCR and amplicon indexing of the V3-V4 region. Individual amplicons were PCR-indexed and pooled. Sequencing was carried out on the Miseq instrument using the 600-cycle MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA). The 4150 TapeStation System (Agilent Technologies, Santa Clara, CA, USA) and NEBNext Library Quant Kit for Illumina (NEB, Ipswich, MA, USA) were used to qualify the mean size and concentration of the DNA libraries. In each sequencing run, the mock community samples (D6305 ZymoBIOMICS Microbial Community DNA Standard, Zymo Research, Irvine, CA, USA) were added to the sample pool.
Bioinformatic analysis
Processing and quality filtering of paired-end reads
A total of 813 samples were analysed, including the mock communities used as controls (ZymoBIOMICS™). The DADA2 software package with default settings was used to process and filter the reads, except for minQ = 6 (Callahan et al., 2016). After filtration, denoising, and merging, 61.4%, 60.8%, and 56.8% of the raw reads were saved; after removing chimeras, 42.1% of the raw reads were saved. The number of amplicons varied in each run. The average number of amplicon sequencing variants (ASVs) was 7,000 per run.
ASV processing
The USEARCH software and UNOISE3 algorithm were used to denoise the amplicons and obtain zero-radius OTUs (ZOTUs) (Edgar and Flyvbjerg, 2015). The ZOTUs were aligned against the 16S-reference database of 22,504 complete 16S rRNA gene sequences from the Genbank (accessed on July 28, 2021) and the representative subset of the Greengenes database (https://www.drive5.com/usearch/manual/download_gg97.html). Chimeras and ‘badly’ aligning ZOTUs were removed, and two clusters were obtained: OTUs99 (clustering identity = 99%) and OTUs97 (clustering identity = 97%) (https://www.drive5.com/usearch/manual/uparse_otu_radius.html). The number of ZOTUs, OTUs99%, and OTUs97% sequences were 4,488, 3,303, and 1,048, respectively.
OTU abundance table
Unfiltered raw reads were merged and pooled using the ‘usearch’ tool with default settings and mapped (60% of the original raw reads) to the ZOTUs and OTUs to create abundance tables. To compare α- and β-diversities in patients and controls, the library size was normalised by rarefying the OUT table to 11,000 sequences per sample using the USEARCH software (v.11.0.667_64).
Taxonomic annotation
Taxa were annotated using the Sintax algorithm, the Ribosomal Database Project’s (RDP) training set of 21,000 sequences (available at https://www.drive5.com/usearch/manual/sintax_downloads.html), and a bootstrap cutoff of 20% (Edgar).
Result reproducibility
To assess reproducibility, run-to-run sequencing of the mock communities was carried out using the ZymoBIOMICS mock community D6305 DNA standard (Zymo Research, Orange County, CA, USA). In most cases, the reproducibility ratio was high. However, with D6300, biases were observed during the DNA isolation of some taxa of gram-positive bacteria: Streptococcus aureus, 50% loss rate; Enterococcus faecium, over 90% loss rate; Listeria, over 90% loss rate. OTUs97% provided the highest reproducibility ratio.
Statistical analysis
Microbial composition and diversity metrics were assessed using sex, age, and antibiotics as covariates. The Scikit-bio 0.5.6 Package (Python 3.8 package) was used to calculate α-diversity metrics. Principal component analysis (PCA) (Python 3.8 package skbio) was used to analyse β-diversity. The first two principal components were used in the regression analysis. The ellipses on the scatter plots show a 2-standard deviation (SD).
To estimate changes in the URT microbiome between the timepoints, log regression coefficients (RCs) were calculated. The significance was assessed using a Z-test.
A network analysis was carried out to analyse correlations between different microbial taxa. The results of the analysis were used to construct a network graph, where each individual taxon is shown as a node and the correlations between them are shown as edges connecting the nodes (based on the Bonferroni-corrected Spearman correlation coefficients). The graph shows only significant taxa (correlation coefficients > 0.3). Low-abundance taxa (relative abundance < 1%) were excluded to avoid false-positive results. The jActiveModules (https://apps.cytoscape.org/apps/jactivemodules, accessed on April 20, 2022) were used to detect clusters within the network.
To remove possible ‘noise’ during the construction of a Venn diagram, a cut-off value of 0.1% was introduced for the mean abundance of taxa in COVID-19 patients and controls.
3 Results
Demographic and clinical characteristics of the participants
This multi-center study followed 285 patients with a median age of 68 years: 126 men, 142 women, and 17 participants who had chosen not to specify their sex or gender. The participants had been admitted to one of the five hospitals in Moscow and Saint Petersburg and had been diagnosed with COVID-19. The median time from symptom onset to hospitalisation was four days (interquartile range (IQR): 2.0-7.0). Moderate COVID-19 was diagnosed in 44.2% of the patients; severe, in 47.0%; and mild, in 8.8%. Table 3 presents the demographic and clinical characteristics of the patients.
Patient characteristics
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
A total of 849 URT samples were collected at three time points (TP): upon hospitalisation (TP0; n = 285); after the median period of 5.0 days after hospitalisation (IQR: 5.0-6.0 days) (TP1; n = 230); and after the median period of 12 days (IQR: 11.0-12.0 days) (TP2; n = 181) after hospitalisation. TP1 and TP2 were at least three days apart but not more than 12 days apart (Figure 1).
Study design: patient enrolment and sample collection.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Upon enrolment, SARS-CoV-2 RNA levels were measured in all patients. Over half of them had an undetectable viral load at the time of testing, despite positive COVID-19 diagnoses. In 47.8% of patients, the viral load was detectable and varied between 1 and 17,760 copies/ml (with a median of 18.0 copies/ml; IQR: 4.8-151.0 copies/ml). Approximately one-third of the patients had normal pulse oximetry readings. Low and critical oxygen saturation were observed in 51.1 and 13.5% of the patients, respectively. Four out of five patients required respiratory support at some point during the treatment period; four patients required intubation. Predictably, serum levels of C-reactive protein (CRP) were significantly elevated in most patients, with a median of 51.2 mg/l (IQR: 19.9-123.5 mg/l).
Table 3 presents the treatment details. The most frequently administered antibiotics were beta-lactams (50.2%) and macrolides (48.8%); 26.5% of the patients were not treated with antibiotics. Other commonly used medications included anticoagulants (23.9%), interferons (13.0%), and IL-6 inhibitors (10.2%). The observed mortality rate was 6%. The rest of the patients recovered and were discharged from the hospital with no follow-up.
A group of 100 healthy individuals with a median age of 60.0 years (IQR: 54.5-65.5) served as controls: 15 men, 68 women, and 17 people who did not specify their sex/gender.
Microbiome composition in patients and controls
Three genera, Prevotella, Streptococcus, and Veillonella, dominated the URT microbial communities in both patients and controls, accounting for over 50% of the URT microbiome (Figure 2, Supplementary Table S3). These were followed by Leptotrichia, Granulicatella, and Rothia in patients and Haemophilus, Neisseria, and Leptotrichia in controls.
Predominant genera in the upper respiratory tract in the patients and controls.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Interestingly, 47 genera were present only in patients, while only 2 were exclusive to controls (Figure 3, Supplementary Table S4). A total of 86 genera were present in both the patients and controls. The most frequently detected COVID-19-specific genera were Methylobacterium, Lactiplantibacillus, and Schwartzia, found in 19.78, 10.07, and 10.07% of patients, respectively. The most frequently detected genera unique to controls were Aeromonas and Oblitimonas, found in 1.22% and 3.66%, respectively (Supplementary Table S4). The number of COVID-19-specific genera reduced over time: to 46 by TP1 (Supplementary Table S5) and 37 by TP2 (Supplementary Table S6).
Venn diagram: the number of genera unique to the patients (the green circle); to the controls (the blue circle); present in both (the overlap). The list below the diagram names 2 most common genera found only in the patients (the left column) and controls (the right column).
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Microbial richness and diversity of the URT in patients and controls
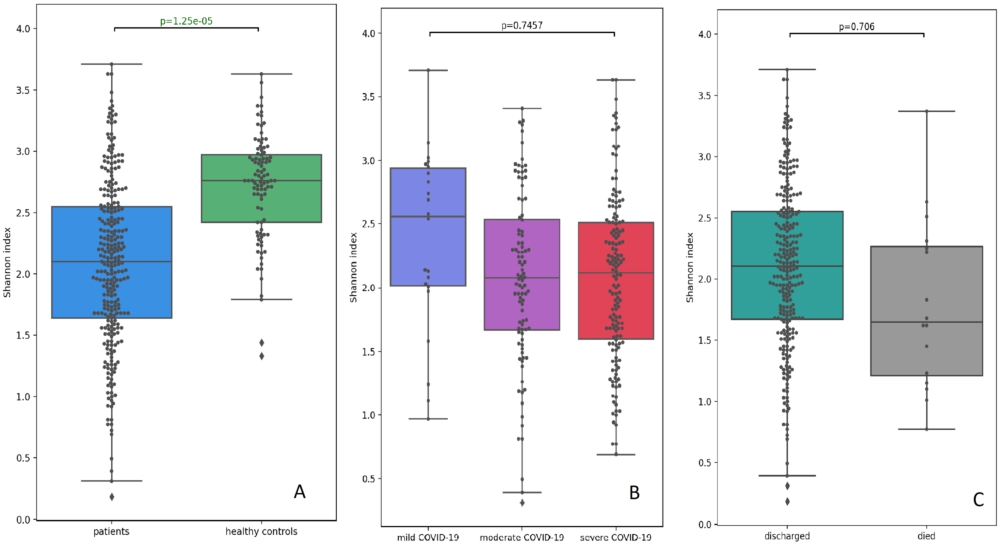
We estimated α-diversity of the URT microbiota based on the Shannon and Chao indices. To compare the diversity metrics in patients and controls, we built a linear regression model using ordinary least squares and adjusted it for age, sex, and antibiotics. Compared with the controls, the patients showed a substantially depleted composition of commensal bacteria, as confirmed by the significantly lower Shannon and Chao indices (
Comparison of the Shannon indices of the upper respiratory tract microbiota between the patients and controls (A); patients with mild, moderate, and severe COVID-19 (B); discharged and deceased patients (C).
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
We also split patients by COVID-19 severity and outcomes and analysed α-diversity in each subgroup. The Shannon and Chao diversity indices in the moderate/severe COVID-19 patients tended to be lower than in mild COVID-19 patients; however, these differences did not reach statistical significance (Figures 4B and Supplementary Figure S1B). Alfa-diversity was not associated with the outcome, fatal or non-fatal; however, the URT microbiota in deceased patients tended to be more markedly depleted (Figures 4C and Supplementary Figure S1C).
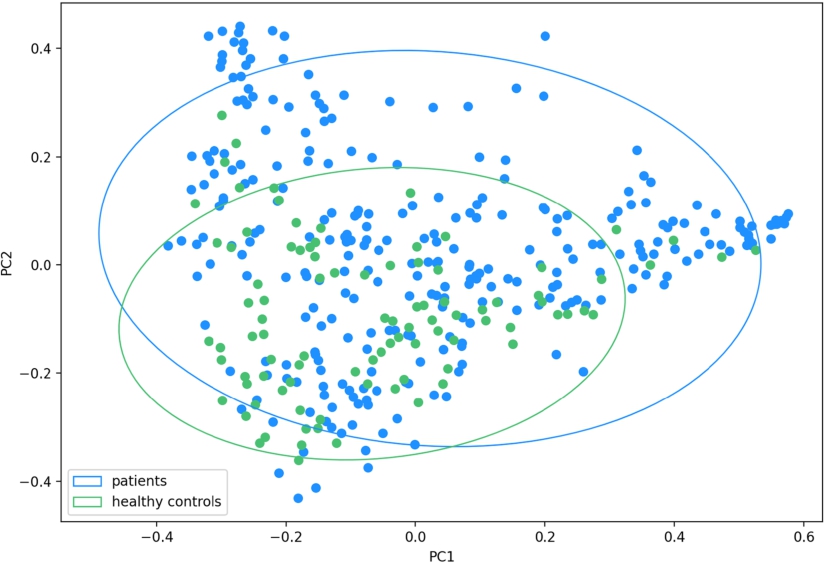
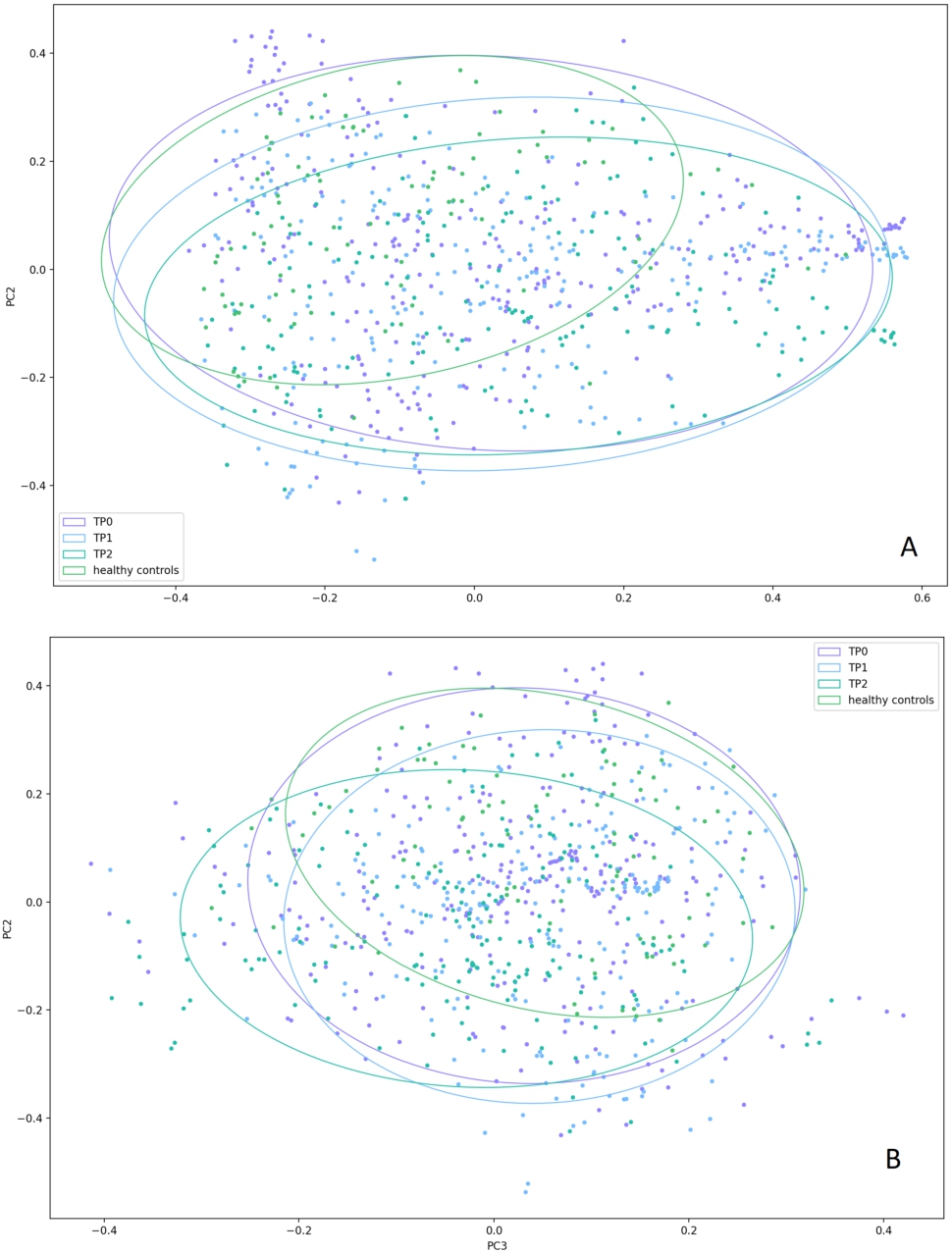
For β-diversity analysis, we used PCA to measure UniFrac distance and Bray-Curtis dissimilarity between URT samples and made adjustments for age, sex, and antibiotic intake. Based on Bray-Curtis dissimilarity, patients and controls formed 2 distinct clusters (PC1,
The Bray-Curtis dissimilarity between the patients’ and controls’ upper respiratory tract microbiota: the microbial composition in the patients and controls differ significantly along both PC1 and PC2 (
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
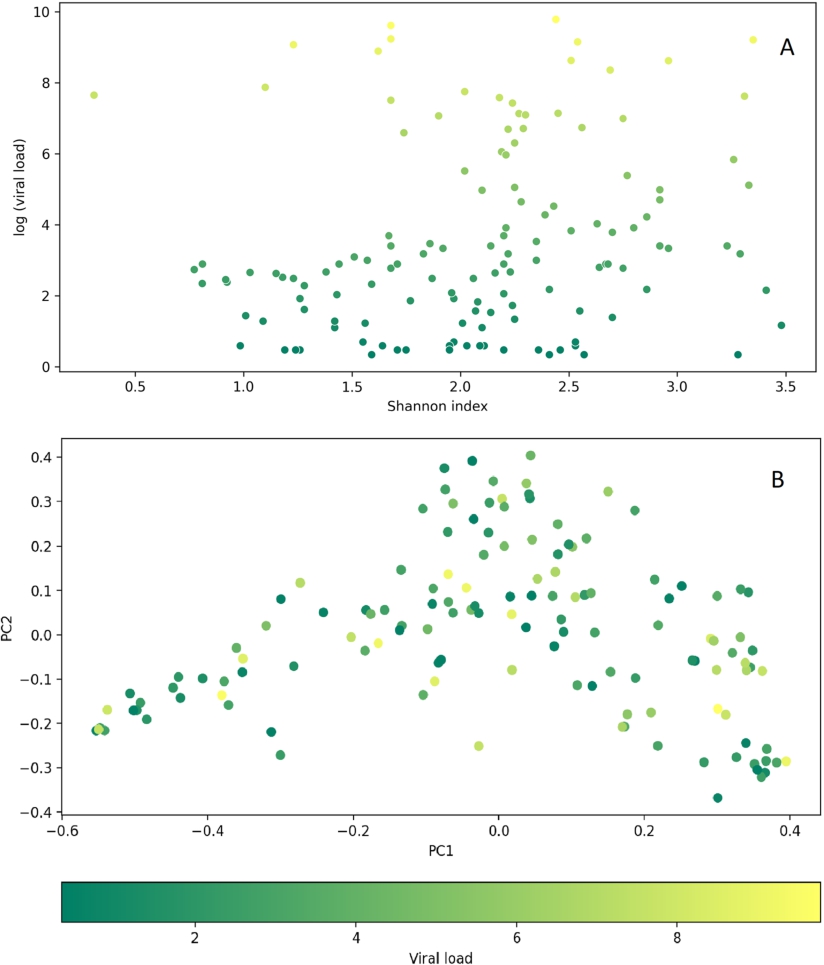
The association analysis of SARS-CoV2 viral load and diversity metrics showed that neither α-diversity (Shannon,
Relationship between SARS-CoV-2 viral load and diversity metrics of the upper respiratory tract microbiota in the COVID-19 patients. (A) The plot showing no association between the Shannon α-diversity indices and the log values of SARS-CoV-2 viral load (
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Differences in the URT microbial communities between the patients and controls
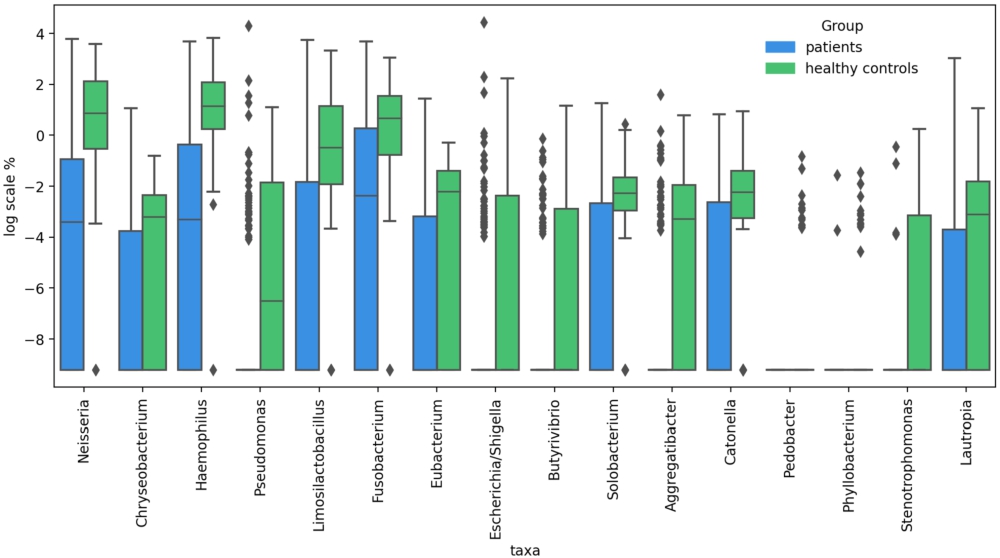
We calculated age- sex-, and antibiotics-adjusted logistic regression coefficients (RCs) to compare the taxonomic enrichment in the URT samples of the patients and controls. Sixteen genera were found to be significantly reduced in the patients compared to controls, including Neisseria (RC = 0.14;
Differentially abundant taxa between COVID-19 patients and healthy individuals.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Neither COVID-19 severity nor treatment outcomes affected the bacterial enrichment in the URT samples; there was no significant difference between the subgroups split by COVID-19 severity or outcomes. No relationship was found between microbial abundance and viral load.
Changes in patients’ URT microbiome
We longitudinally profiled patients’ URT microbiome throughout their hospital stay to evaluate short-term changes. All calculations were adjusted for age, sex, and antibiotics.
We observed a minor loss in α-diversity at TP1 compared with the baseline, followed by a minor increase at TP2 compared with TP1; however, none of these changes reached statistical significance. We obtained
The dynamics of Shannon indices in the COVID-19 patients during their treatment. TP0: time-point 0 (upon admission to the hospital); TP1: time-point 1 (after a median of 5.0 days); TP2: time-point 2 (after a median of 12.0 days).
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Then, we sought to evaluate how patients’ URT microbiome changed between the timepoints. Based on the Bray-Curtis dissimilarity, both along PC2 and PC3, TP3 samples were more similar to controls (
The Bray-Curtis dissimilarity between the upper respiratory tract microbiota in the patient samples collected at different time-points during their treatment and control samples. (A) the Bray-Curtis distances plotted along PC1 and PC2. Significant differences were observed only along PC2 between the TP0 samples and the control samples (
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
We also used the Bray-Curtis dissimilarity matrix to estimate the inter-sample variability in patients’ samples collected at different timepoints and compare them with that of controls. We measured standard deviations (SDs) of the samples along PC1 and PC2 to estimate the dispersion and measured mean distances from the samples to the centre of one of the four clusters they belong to (healthy controls, patients at TP0, TP1, or TP2). The highest dispersion was observed in the patients at TP0 (SD along PC1, 0.26; SD along PC2, 0.20), but it tended to decrease by TP1 (SD along PC1, 0.26; SD along PC2, 0.17) and TP2 (SD along PC1, 0.25; SD along PC2, 0.14). The healthy controls demonstrated the lowest dispersion (SD along PC1, 0.20; SD along PC2, 0.14). The highest mean distance to the centre of the cluster was also observed in the patients at TP0 (0.29), which was significantly higher than that in the controls (0.21) (
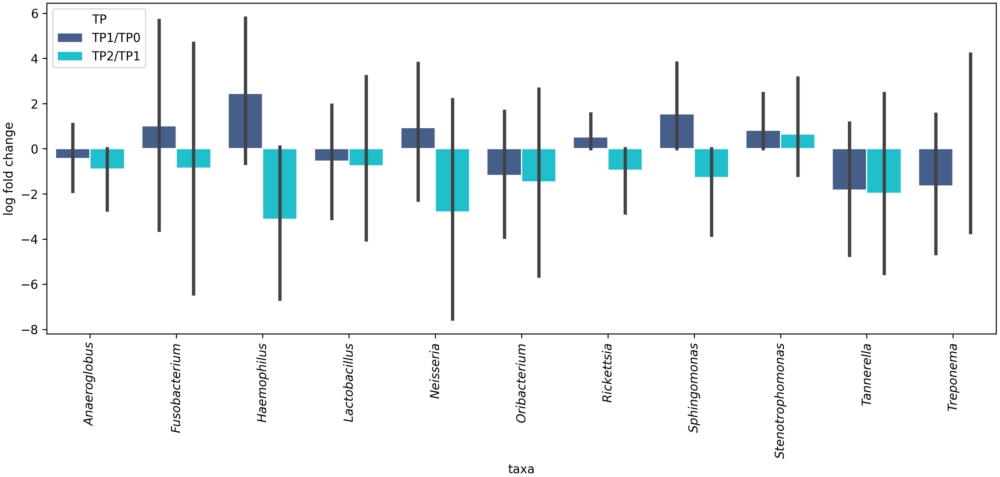
To estimate differential microbial abundance at different time-points, we calculated RCs with unadjusted and Bonferroni-adjusted P-values. Five genera, including Anaeroglobus (RC = 0.49;
Inter-time-point changes in the relative abundance of the upper respiratory tract microorganisms in the COVID-19 patients during their treatment. TP1/TP0 – change registered at time-point 1 (after a median of 5.0 days following admission to the hospital) compared with time-point 0 (upon admission to the hospital). TP2/TP1 – change registered at time-point 2 (after a median of 12.0 days following admission to the hospital) compared with time-point 1 (after a median of 5.0 days following admission to the hospital). Red frames show significant changes.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
We also attempted to examine microbial abundance changes in deceased patients. We tracked changes in those genera, the abundance of which had changed between the time points in all patients. Interestingly, this subgroup showed a tendency toward a decrease in the abundance of numerous taxa, such as Anaeroglobus, Fusobacterium, Haemophilus, Lactobacillus, Oribacterium, Neisseria, Sphingomonas, and Tannerella, that had become more abundant by TP2 in the entire cohort (Figure 11).
Inter-time-point changes in the relative abundance of the upper respiratory tract microorganisms in the deceased COVID-19 patients during their treatment. TP1/TP0 – change registered at time-point 1 (after a median of 5.0 days following admission to the hospital) compared with time-point 0 (upon admission to the hospital). TP2/TP1 – change registered at time-point 2 (after a median of 12.0 days following admission to the hospital) compared with time-point 1 (after a median of 5.0 days following admission to the hospital). None of the changes were significant.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Network analysis of the microbial communities in the patients and controls
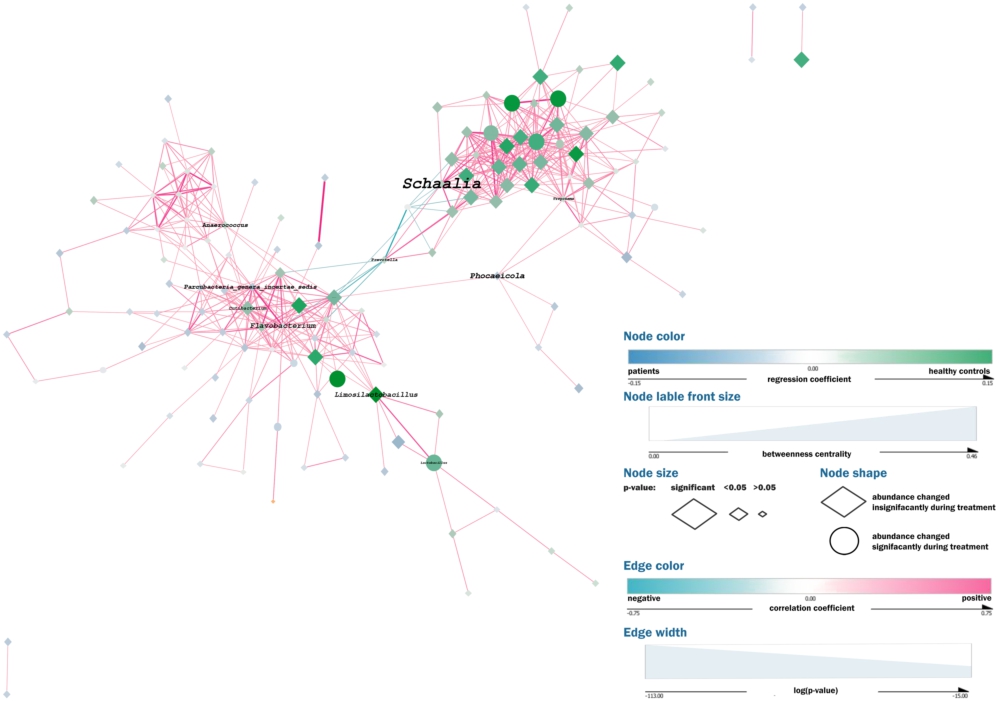
We constructed a co-occurrence network using metagenomic samples from 285 COVID-19 patients and 100 healthy controls to analyse correlations between different microbial taxa (Figure 12; Interactive Supplement 2). The graph also shows the differences in the bacterial abundance between patients and controls, as well as the previously calculated abundance changes in patients during the treatment (see sections ‘Differences in the URT microbial communities between patients and controls’ and ‘Changes in patients’ URT microbiome’).
Network analysis. Each node in the network represents an individual taxon with its colour reflecting the difference in the relative abundance of this taxon between COVID-19 patients and healthy controls (blue – overrepresented in the patients; green – overrepresented in controls) and its size proportional to the significance of this difference. The shape of the node shows whether the abundance of the corresponding taxon has changed during treatment. The edges denote positive (pink) or (negative) correlations between the taxa, while the edges’ width is proportional to the significance of the correlation.
Citation: Beneficial Microbes 15, 2 (2024) ; 10.1163/18762891-bja00004
Each node in the network represents an individual taxon. The colour reflects the difference in the taxon’s relative abundance between COVID-19 patients and healthy controls (blue for overrepresented in patients; green for overrepresented in controls); the size of the node is proportional to the significance of the difference; and the shape shows whether the abundance of the taxon has changed during the treatment. The edges denote positive (pink) or negative correlations between the taxa, while their width is proportional to the significance of the correlation.
The network has 137 nodes, arranged in one major and three minor independent clusters, and 556 edges. The average number of neighbouring nodes is 8.28 per each node. The network density is 0.064; network diameter is 9; the clustering coefficient is 0.49. The network centralisation is 0.14; the characteristic path length is 3.89.
Most correlations between the bacterial taxa were positive. The Neisseria-Haemophilus – Fusobacterium triangle (that had emerged as the taxa more typical of the healthy controls) showed some of the strongest relationships in the major cluster of the network.
All the negative correlations in the network involved Streptococcus and Prevotella, with the strongest negative correlation formed between these two taxa.
4 Discussion
In our study, we compared the URT microbiome in COVID-19 patients and healthy controls. The COVID-19 patients demonstrated distinctive URT microbiome and dynamics. The Bray-Curtis dissimilarity showed that their URT microbiota differed dramatically from those of the healthy controls. SARS-CoV-2 infection was associated with a severe URT dysbiosis and loss of microbial diversity, as evidenced by lower Shannon and Chao indices in the COVID-19 patients. Moreover, we observed a trend toward lower α-diversity in the patients with more severe COVID-19 and in deceased patients, suggesting an aggravation of URT dysbiosis along with disease progression. To minimise the effects of such variables as age, gender, and antibiotics, all results were adjusted accordingly. Thus, the observed differences support the hypothesis that SARS-CoV-2 alone might cause changes in the URT microbiome.
The URT microbiome in the healthy controls demonstrated a substantially higher α-diversity. However, in their URT specimens, we observed only two unique genera as opposed to 47 genera exclusively found in the COVID-19 patients, indicating that microbiome dysbiosis differs on case by case basis. Specifically, microbiomes disturbed by stress factors tend to change in a multitude of different ways. We believe that this vast number of atypical taxa in our cohort of patients confirms the destabilising effect of COVID-19 on the URT microbiome.
We hypothesise that these 47 COVID-19-specific genera could have emerged due to the microbiome instability caused by SARS-CoV-2, since, unlike a healthy URT microbiome, a compromised one is more prone to stochastic compositional shifts and ‘occasional acquisitions’ of new taxa. This hypothesis is consistent with the fact that the patients’ URT specimens collected upon admission to the hospital demonstrated a significantly higher dispersion than that of controls, suggesting higher inter-individual variation that can be interpreted as a marker of microbiome instability. The disappearance of some COVID-19-specific genera by TP1 and TP2 appears to indicate a trend towards a more stabilised URT ecosystem during treatment. Again, this is in line with our finding suggesting a gradual reduction in the dispersion of patients’ specimens over time, which is likely to confirm increasing microbiome stability.
Poor oral hygiene and oxygen support in the COVID-19 patients might have contributed to the emergence of new genera in their URT specimens. Indeed, some of these taxa were found to be associated with oral cavity diseases. For example, Scardovia was previously reported in patients with caries (McDaniel et al., 2021), as well as in COVID-19 patients admitted to intensive care units (Rueca et al., 2021). Schwartzia was associated with periodontal disease (Sisk-Hackworth et al., 2021), while Olsenella was detected in dentine samples from patients with irreversible pulpitis (Selvakumar et al., 2021). In addition, several taxa identified as COVID-19-specific in our study, such as Methylobacterium, Serratia, Salmonella, and Moraxella, can potentially include pathogenic species capable of causing serious infections, particularly in an immunocompromised host. This is in line with the hypothesis that COVID-19 patients are prone to bacterial superinfections that often cause the aggravation of pneumonia and an increased the risk of death (Zhou et al., 2020).
We should take into consideration that the COVID-19 patients, unlike healthy controls, were in a specific hospital environment, where they were exposed to multiple microorganisms not usually found elsewhere. Moreover, it is extremely challenging to differentiate between the core microbiome species and transient ones, given the constant gas exchange between the airways and the environment. Therefore, some of these taxa acquired by the patients can be transient, especially those with a low abundance. Nevertheless, the reduction in the number of COVID-19-specific taxa by the end of the treatment (when patients were still in hospital), as well as decreasing stochasticity might signal that the URT microbiome trends towards stabilisation, somehow trying to resist the acquisition and ‘anchorage’ of the new species from the environment.
Studies on the microbial diversity in COVID-19 patients have reported different findings. Some reported a lower microbial diversity in COVID-19 patients than in healthy controls (Ren et al., 2021; Soffritti et al., 2021), while others found no such correlation (De Maio et al., 2020; Miller et al., 2021). Many authors reported a significantly diminished richness of species in patients with increasingly severe disease symptoms (Ma et al., 2021; Soffritti et al., 2021) or in deceased patients (Ventero et al., 2022). Meanwhile, other studies reported a higher bacterial richness and diversity in COVID-19 patients than in controls (Rosas-Salazar et al., 2021) or an increased diversity in response to increasing disease severity (Shilts et al., 2021). On the one hand, these conflicting findings might be due to differences in the study designs, laboratory testing techniques, data analysis, etc. On the other hand, they could be explained by an extreme instability of the diseased microbiome that manifests itself in a higher α-diversity in COVID-19 patients or in patients with more severe COVID-19 symptoms, although one would expect the opposite.
Therefore, increased microbial diversity might not always be an indicator of a healthier microbiome and should be interpreted with caution. Most authors, nonetheless, agree that the SARS-CoV-2 infection results in a significant URT dysbiosis. Our findings corroborate and build upon the results of earlier studies.
The abundance of 16 genera significantly decreased in the COVID-19 patients upon their admission to the hospital compared with the healthy controls. Some of these genera have already drawn the attention of other researchers who analysed the airway microbiome in COVID-19. The depletion of Neisseria and/or Haemophilus in COVID-19 patients has been reported by several authors (Iebba et al., 2021; Rosas-Salazar et al., 2021). De Castilhos et al. found that a lower abundance of Neisseria subflava and Haemophilus spp. was associated with an increased risk of mortality (de Castilhos et al., 2022). Merenstein et al. reported significantly reduced levels of Neisseria and Haemophilus in patients with more severe COVID-19 symptoms, whereas in a healthy URT microbiome, these bacteria are usually abundant (Merenstein et al., 2022). Pozzi et al. (2022) showed that Haemophilus was underrepresented in hospitalised COVID-19 patients compared with outpatients. In the study by Nardelli et al. (2021), Fusobacterium periodonticum was one of the most markedly reduced microorganisms in COVID-19 patients compared with controls. Fusobacterium depletion in COVID-19 patients, compared with healthy individuals, was also reported by Ren et al. (2021).
It is challenging to conclusively determine the causes of the depletion of each individual taxon observed in our cohort of COVID-19 patients, especially given the multitude of relationships between the URT-inhabiting microorganisms, as demonstrated by the network analysis. Most probably, the changes could be attributed to the general URT dysbiosis caused by SARS-CoV-2, which disrupts the interactions between the commensal bacteria. Nonetheless, exact reasons could be hypothesised, at least for some of these microorganisms. For example, non-pathogenic Neisseria species are known to prevent colonisation of the URT mucosa by pathogenic microorganisms (Dorey et al., 2019) and are thought to modulate the local innate immune response (Powell et al., 2018).
We also sought to evaluate short-term changes in the patients’ microbiome during their in-hospital stay. We performed a metagenomic analysis of the URT specimens obtained after a median of 5 days (TP1) and 12 days (TP2) since the admission to the hospital and compared them to the baseline specimens. Alpha-diversity did not change significantly and remained at the same level throughout the entire study period, presumably because the URT microbial community cannot be fully restored within such a short period of time.
However, there were some rapid changes in the taxonomic composition of the specimens by the end of the in-hospital stay. First and foremost, there was a significant increase in the abundance of six genera, including Fusobacterium, Haemophilus, Neisseria, Rickettsia, Sphingomonas, and Stenotrophomonas. This finding is particularly interesting because four of them were differentially abundant between the patients at TP0 and the controls: Fusobacterium, Haemophilus, Neisseria, and Stenotrophomonas. This suggests that the URT microbial composition of COVID-19 patients not only changes during the treatment but also approaches that of healthy controls. Such an increase in the genera overrepresented in a healthy URT could be interpreted as a sign of microbiome stabilisation and ‘healing’. One might attribute this positive dynamic to the treatment, such as the administration of antibiotics. However, antibiotics are known to cause microbial disbalance, rendering some beneficial microorganisms collateral damage (Patangia et al., 2022) Their administration alters the URT microbiome, leading to dysbiosis and the colonisation of the URT by opportunistic and resistant pathogens (Pérez-Cobas et al., 2022). Therefore, it is highly improbable that these positive changes were due to the treatment. Moreover, the abundance of three of these four ‘healthy’ URT genera (Fusobacterium, Haemophilus, Neisseria) decreased, although not significantly, in the patients who died after TP1, probably suggesting the aggravation of URT dysbiosis.
The analysis of Bray-Curtis dissimilarity between the specimens collected from the patients at three different timepoints and controls provided further evidence of a rapid URT microbiome ‘healing’. The patients’ samples at TP2 (end of treatment) were compositionally significantly closer to the control samples than to the patients’ samples at TP0 (baseline). Thus, the COVID-19 patients in our study clearly demonstrated a certain degree of normalisation of the URT microbiome soon after their admission to the hospital, despite the fact that many of them were treated with antibiotics.
This study has several limitations. Most patients had moderate to severe COVID-19; very few had mild COVID-19; hence, the cohort was relatively homogenous. Individuals with minor COVID-19 symptoms are usually treated in outpatient settings; therefore, they are not represented in our study that focused on hospitalised individuals. Due to difficulties with enrolment and ethical considerations, there were very few critically ill patients in this study. Thus, the absence of mild and extremely severe COVID-19 cases might have prevented us from finding significant clustering of the URT samples by disease severity and outcome. There was no post-discharge follow-up due to a number of technicalities; hence, we could not analyse long-term outcomes, survival, or further changes in the patients’ URT microbiota. A follow-up might have provided valuable information on the long-term dynamics and prognosis and facilitated a better discrimination between the core and transient – hospital-specific – taxa.
All the patients underwent treatment with medications, including antiviral and/or antibacterial drugs. The treatment certainly affected their URT microbiome, especially antibiotics. Antibacterial therapy was used as a covariate; however, we cannot definitively conclude it had no effect on our findings. With the given study design, it is almost impossible to specifically distinguish between the effects of SARS-CoV-2 and those of numerous confounders, such as pharmacotherapy, respiratory support, a specific hospital environment, immune shifts in the airway mucosa, etc. Further studies should warrant a more detailed insight into the COVID-19-associated URT microbiome and a better understanding of interactions between SARS-CoV-2 and airway microbial communities.
Lastly, given the relative stability of the human microbiome, the URT microbiome of the controls was sampled only at the initial time-point and was used as a reference for all time-points in COVID-19 patients. Although it is affected by a range of factors, such as the environment, lifestyle, age, and others, a healthy human microbiome is known to be relatively stable. Studies have shown that it can remain unchanged for years (Faith et al., 2013). Therefore, no further sample collection of the control URT microbiomes was carried out after the initial sampling. Tracking the naturally occurring alterations in a healthy URT microbiome was outside the scope of the present research, which focused on specifically analysing the URT microbiome in moderately and severely ill COVID-19 patients and tracking their recovery dynamics.
Our work has several strengths, one of which is a relatively large sample size. With a substantial number of both COVID-19 patients recruited at different hospitals and controls, we were able to provide more accurate and reliable findings on the URT microbiome and its association with the SARS-CoV-2 infection. Furthermore, we made proper adjustments to minimise the confounding effect of several covariates, including age, sex, and antibiotics, which are known to affect the microbial composition but were often insufficiently analysed in previously published studies. We tried to address this issue by conducting a multivariate analysis using the appropriate covariates. Another strength of our study is its longitudinal design, which allowed us to trace the dynamics of microbial changes occurring shortly after the patient’s admission to the hospital. The majority of studies on this subject were cross-sectional and did not assess changes in the URT microbial communities over time, although they might have given valuable information about the processes occurring in the URT.
5 Conclusions
In our study, the SARS-CoV-2 infection was associated with a significant destabilisation of the URT microbiome in hospitalised patients. The infection had a particularly serious impact on several taxa. SARS-CoV-2 is a strong stressor and appears to increase the URT microbiome’s susceptibility to occasional stochastic changes that result in general destabilisation. This destabilisation might be a predisposing factor for bacterial and/or viral superinfections, especially in the hospital setting, where there is a high probability of nosocomial transmission of pathogens. Nevertheless, the URT microbiome demonstrated resilience against stochastic changes and the capacity to ‘undo’ the damage over time, as shown by the rapid restoration of a ‘healthier’ composition and decreasing stochasticity soon after hospitalisation.
Corresponding author; e-mail: oignatieva@cspfmba.ru
Supplementary material
Supplementary material is available online at: https://doi.org/10.6084/m9.figshare.25144397
Supplementary material S1. Case report form for COVID-19 patients.
Table S1. ddPCR reaction mixture.
Table S2. Amplification protocol for ddPCR.
Table S3. Most abundant genera in the upper respiratory tract microbiome of the COVID-19 patients and controls.
Table S4. Unique representatives of the upper respiratory tract microbiome in the COVID-19 patients upon their admission to hospital (at TP0) and controls.
Table S5. Unique and representatives of the upper respiratory tract microbiome in the COVID-19 patients at TP1 and controls.
Table S6. Unique representatives of the upper respiratory tract microbiome in the COVID-19 patients at TP2 and controls.
Table S7. Inter-time-point changes in the relative abundance of various upper respiratory tract microorganisms in the COVID-19 patients.
Figure S1. Comparison of the Chao indices of the upper respiratory tract microbiota between the patients and controls (A); patients with mild, moderate, and severe COVID-19 (B); discharged and deceased patients (C).
Figure S2. The Unifrac distances between the patients’ and controls’ upper respiratory tract microbiota.
Figure S3. The Bray-Curtis dissimilarity between the upper respiratory tract microbiota of the patients with mild, moderate, and severe COVID-19 (A) and discharged or deceased patients (B).
Figure S4. Unifrac distances of the upper respiratory tract microbiota in patients with mild, moderate, and severe COVID-19 pneumonia (A), and patients discharged from hospital or died (B).
Figure S5. Relationship between SARS-CoV-2 viral load and diversity metrics of the upper respiratory tract microbiota in the COVID-19 patients.
Figure S6. The dynamics of Chao indices in the COVID-19 patients during their treatment.
Figure S7. Changes in the Shannon indices of the upper respiratory tract microbiota in the patients with mild (A), moderate (B), and severe (C) COVID-19 throughout their treatment.
Figure S8. Changes in the Chao indices of the upper respiratory tract microbiota in the patients with mild (A), moderate (B), and severe (C) COVID-19 throughout their treatment.
Figure S9. Changes in the Shannon indices of the upper respiratory tract microbiota in the discharged (A) and deceased (B) patients.
Figure S10. Changes in the Chao indices of the upper respiratory tract microbiota in the discharged (A) and deceased (B) patients.
Figure S11. The Unifrac distances between the upper respiratory microbiota in the patient samples collected at different time-points during their treatment and control samples.
Interactive Supplement 1 and 2.
Authors’ contribution
All authors contributed equally to the writing of this paper. All authors read and approved the final manuscript.
Conflict of interest
The authors have declared no conflict of interest.
References
Borges, L., Giongo, A., Pereira, L.M., Trindade, F.J., Gregianini, T.S., Campos, F.S., Ghedin, E. and da Veiga, A.B.G., 2018. Comparison of the nasopharynx microbiome between influenza and non-influenza cases of severe acute respiratory infections: a pilot study. Health Science Reports 1: e47. https://doi.org/10.1002/hsr2.47
Braun, T., Halevi, S., Hadar, R., Efroni, G., Glick Saar, E., Keller, N., Amir, A., Amit, S. and Haberman, Y., 2021. SARS-CoV-2 does not have a strong effect on the nasopharyngeal microbial composition. Scientific Reports 11: 8922. https://doi.org/10.1038/s41598-021-88536-6
Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J. and Holmes, S.P., 2016. DADA2: high-resolution sample inference from illumina amplicon data. Nature Methods 13: 581-583. https://doi.org/10.1038/nmeth.3869
de Castilhos, J., Zamir, E., Hippchen, T., Rohrbach, R., Schmidt, S., Hengler, S., Schumacher, H., Neubauer, M., Kunz, S., Muller-Esch, T., Hiergeist, A., Gessner, A., Khalid, D., Gaiser, R., Cullin, N., Papagiannarou, S.M., Beuthien-Baumann, B., Kramer, A., Bartenschlager, R., Jager, D., Muller, M., Herth, F., Duerschmied, D., Schneider, J., Schmid, R.M., Eberhardt, J.F., Khodamoradi, Y., Vehreschild, M., Teufel, A., Ebert, M.P., Hau, P., Salzberger, B., Schnitzler, P., Poeck, H., Elinav, E., Merle, U. and Stein-Thoeringer, C.K., 2022. Severe dysbiosis and specific Haemophilus and Neisseria signatures as hallmarks of the oropharyngeal microbiome in critically ill coronavirus disease 2019 (COVID-19) patients. Clinical Infectious Diseases 75: e1063-e1071. https://doi.org/10.1093/cid/ciab902
De Maio, F., Posteraro, B., Ponziani, F.R., Cattani, P., Gasbarrini, A. and Sanguinetti, M., 2020. Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biological Procedures Online 22: 18. https://doi.org/10.1186/s12575-020-00131-7
Dorey, R.B., Theodosiou, A.A., Read, R.C. and Jones, C.E., 2019. The nonpathogenic commensal Neisseria: friends and foes in infectious disease. Current Opinion in Infectious Diseases 32: 490-496. https://doi.org/10.1097/QCO.0000000000000585
Edgar, R.C., 2016. SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv (unpeerrevied). https://doi.org/10.1101/074161
Edgar, R.C. and Flyvbjerg, H., 2015. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31: 3476-3482. https://doi.org/10.1093/bioinformatics/btv401
Engen, P.A., Naqib, A., Jennings, C., Green, S.J., Landay, A., Keshavarzian, A. and Voigt, R.M., 2021. Nasopharyngeal microbiota in SARS-CoV-2 positive and negative patients. Biological Procedures Online 23: 10. https://doi.org/10.1186/s12575-021-00148-6
Faith, J.J., Guruge, J.L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A.L., Clemente, J.C., Knight, R., Heath, A.C., Leibel, R.L., Rosenbaum, M. and Gordon, J.I., 2013. The long-term stability of the human gut microbiota. Science 341: 1237439. https://doi.org/10.1126/science.1237439
Grier, A., Gill, A.L., Kessler, H.A., Corbett, A., Bandyopadhyay, S., Java, J., Holden-Wiltse, J., Falsey, A.R., Topham, D.J., Mariani, T.J., Caserta, M.T., Walsh, E.E. and Gill, S.R., 2021. Temporal dysbiosis of infant nasal microbiota relative to respiratory syncytial virus infection. Journal of Infectious Diseases 223: 1650-1658. https://doi.org/10.1093/infdis/jiaa577
Iebba, V., Zanotta, N., Campisciano, G., Zerbato, V., Di Bella, S., Cason, C., Luzzati, R., Confalonieri, M., Palamara, A.T. and Comar, M., 2021. Profiling of oral microbiota and cytokines in COVID-19 patients. Frontiers in Microbiology 12: 671813. https://doi.org/10.3389/fmicb.2021.671813
Kloepfer, K.M., Sarsani, V.K., Poroyko, V., Lee, W.M., Pappas, T.E., Kang, T., Grindle, K.A., Bochkov, Y.A., Janga, S.C., Lemanske, R.F., Jr. and Gern, J.E., 2017. Community-acquired rhinovirus infection is associated with changes in the airway microbiome. Journal of Allergy and Clinical Immunology 140: e318, 312-315. https://doi.org/10.1016/j.jaci.2017.01.038
Ma, S., Zhang, F., Zhou, F., Li, H., Ge, W., Gan, R., Nie, H., Li, B., Wang, Y., Wu, M., Li, D., Wang, D., Wang, Z., You, Y. and Huang, Z., 2021. Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduction and Targeted Therapy 6: 191. https://doi.org/10.1038/s41392-021-00614-3
Man, W.H., de Steenhuijsen Piters, W.A. and Bogaert, D., 2017. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nature Review Microbiology 15: 259-270. https://doi.org/10.1038/nrmicro.2017.14
McDaniel, S., McDaniel, J., Howard, K.M. and Kingsley, K., 2021. Molecular screening and analysis reveal novel oral site-specific locations for the cariogenic pathogen Scardovia wiggsiae. Dentistry Journal 9: 73. https://doi.org/10.3390/dj9060073
Merenstein, C., Bushman, F.D. and Collman, R.G., 2022. Alterations in the respiratory tract microbiome in COVID-19: current observations and potential significance. Microbiome 10: 165. https://doi.org/10.1186/s40168-022-01342-8
Miller, E.H., Annavajhala, M.K., Chong, A.M., Park, H., Nobel, Y.R., Soroush, A., Blackett, J.W., Krigel, A., Phipps, M.M., Freedberg, D.E., Zucker, J., Sano, E.D., Uhlemann, A.C. and Abrams, J.A., 2021. Oral microbiome alterations and SARS-CoV-2 saliva viral load in patients with COVID-19. Microbiology Spectrem 9: e0005521. https://doi.org/10.1128/Spectrum.00055-21
Mostafa, H.H., Fissel, J.A., Fanelli, B., Bergman, Y., Gniazdowski, V., Dadlani, M., Carroll, K.C., Colwell, R.R. and Simner, P.J., 2020. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. mBio 11. https://doi.org/10.1128/mBio.01969-20
Nardelli, C., Gentile, I., Setaro, M., Di Domenico, C., Pinchera, B., Buonomo, A.R., Zappulo, E., Scotto, R., Scaglione, G.L., Castaldo, G. and Capoluongo, E., 2021. Nasopharyngeal microbiome signature in COVID-19 positive patients: can we definitively get a role to Fusobacterium periodonticum? Frontiers in Cellular and Infection Microbiology 11: 625581. https://doi.org/10.3389/fcimb.2021.625581
Powell, D.A., Ma, M., So, M. and Frelinger, J.A., 2018. The commensal Neisseria musculi modulates host innate immunity to promote oral colonization. Immunohorizons 2: 305-313. https://doi.org/10.4049/immunohorizons.1800070
Pozzi, C., Levi, R., Braga, D., Carli, F., Darwich, A., Spadoni, I., Oresta, B., Dioguardi, C.C., Peano, C., Ubaldi, L., Angelotti, G., Bottazzi, B., Garlanda, C., Desai, A., Voza, A., Azzolini, E., Cecconi, M., Mantovani, A., Penna, G., Barbieri, R., Politi, L.S. and Rescigno, M., ICH COVID-19 Task-force, 2022. A ‘multiomic’ approach of saliva metabolomics, microbiota, and serum biomarkers to assess the need of hospitalization in coronavirus disease 2019. Gastro Hep Advances 1: 194-209. https://doi.org/10.1016/j.gastha.2021.12.006
Ren, Z., Wang, H., Cui, G., Lu, H., Wang, L., Luo, H., Chen, X., Ren, H., Sun, R., Liu, W., Liu, X., Liu, C., Li, A., Wang, X., Rao, B., Yuan, C., Zhang, H., Sun, J., Chen, X., Li, B., Hu, C., Wu, Z., Yu, Z., Kan, Q. and Li, L., 2021. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 70: 1253-1265. https://doi.org/10.1136/gutjnl-2020-323826
Rosas-Salazar, C., Kimura, K.S., Shilts, M.H., Strickland, B.A., Freeman, M.H., Wessinger, B.C., Gupta, V., Brown, H.M., Rajagopala, S.V., Turner, J.H. and Das, S.R., 2021. SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. Journal of Allergy and Clinical Immunology 147: e1222, 1226-1233. https://doi.org/10.1016/j.jaci.2021.02.001
Rueca, M., Fontana, A., Bartolini, B., Piselli, P., Mazzarelli, A., Copetti, M., Binda, E., Perri, F., Gruber, C.E.M., Nicastri, E., Marchioni, L., Ippolito, G., Capobianchi, M.R., Di Caro, A. and Pazienza, V., 2021. Investigation of nasal/oropharyngeal microbial community of COVID-19 patients by 16S rDNA sequencing. International Journal of Environmental Research and Public Health 18: 2174. https://doi.org/10.3390/ijerph18042174
Selvakumar, D.R., Krishnamoorthy, S., Venkatesan, K., Ramanathan, A., Abbott, P.V. and Angambakkam Rajasekaran, P., 2021. Active bacteria in carious dentin of mandibular molars with different pulp conditions: an in vivo study. Journal of Endodontics 47: 1883-1889. https://doi.org/10.1016/j.joen.2021.08.018
Shilts, M.H., Rosas-Salazar, C., Strickland, B.A., Kimura, K.S., Asad, M., Sehanobish, E., Freeman, M.H., Wessinger, B.C., Gupta, V., Brown, H.M., Boone, H.H., Patel, V., Barbi, M., Bottalico, D., O’Neill, M., Akbar, N., Rajagopala, S.V., Mallal, S., Phillips, E., Turner, J.H., Jerschow, E. and Das, S.R., 2021. Severe COVID-19 is associated with an altered upper respiratory tract microbiome. Frontiers in Cellular and Infection Microbiology 11: 781968. https://doi.org/10.3389/fcimb.2021.781968
Sisk-Hackworth, L., Ortiz-Velez, A., Reed, M.B. and Kelley, S.T., 2021. Compositional data analysis of periodontal disease microbial communities. Frontiers in Microbiology 12: 617949. https://doi.org/10.3389/fmicb.2021.617949
Soffritti, I., D’Accolti, M., Fabbri, C., Passaro, A., Manfredini, R., Zuliani, G., Libanore, M., Franchi, M., Contini, C. and Caselli, E., 2021. Oral microbiome dysbiosis is associated with symptoms severity and local immune/inflammatory response in COVID-19 patients: a cross-sectional study. Frontiers in Microbiology 12: 687513. https://doi.org/10.3389/fmicb.2021.687513
Ventero, M.P., Moreno-Perez, O., Molina-Pardines, C., Paytuvi-Gallart, A., Boix, V., Escribano, I., Galan, I., Gonzalez-delaAleja, P., Lopez-Perez, M., Sanchez-Martinez, R., Merino, E. and Rodriguez, J.C., 2022. Nasopharyngeal microbiota as an early severity biomarker in COVID-19 hospitalised patients. Journal of Infection 84: 329-336. https://doi.org/10.1016/j.jinf.2021.12.030
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., Xiang, J., Wang, Y., Song, B., Gu, X., Guan, L., Wei, Y., Li, H., Wu, X., Xu, J., Tu, S., Zhang, Y., Chen, H. and Cao, B., 2020. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet 395: 1054-1062. https://doi.org/10.1016/S0140-6736(20)30566-3







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}